About MorphoRegions
The MorphoRegions package is built to computationally identify regions (morphological, functional, etc.) in serially homologous structures such as, but not limited to, the vertebrate backbone. Regions are modeled as segmented linear regressions with each segment corresponding to a region and region boundaries (or breakpoints) corresponding to changes along the serially homologous structure. The optimal number of regions and their breakpoint positions are identified without a priori assumptions. The algorithm fits increasingly complex models (increasing number of regions). For each given number of regions, all possible combinations of breakpoint positions will be iteratively fitted. Maximum-likelihood methods are then used to identify the best model, as described in Gillet et al. (2024).
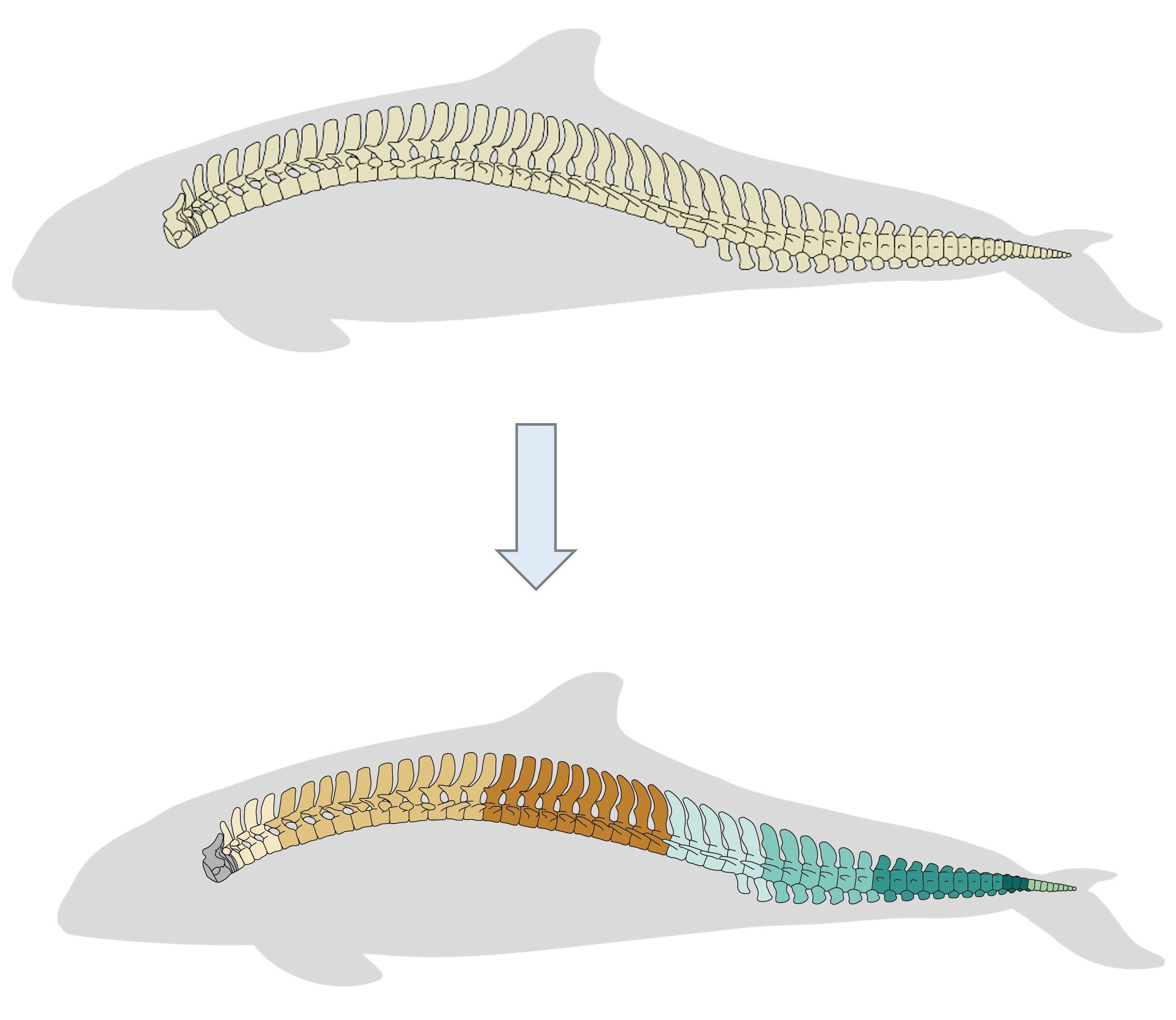
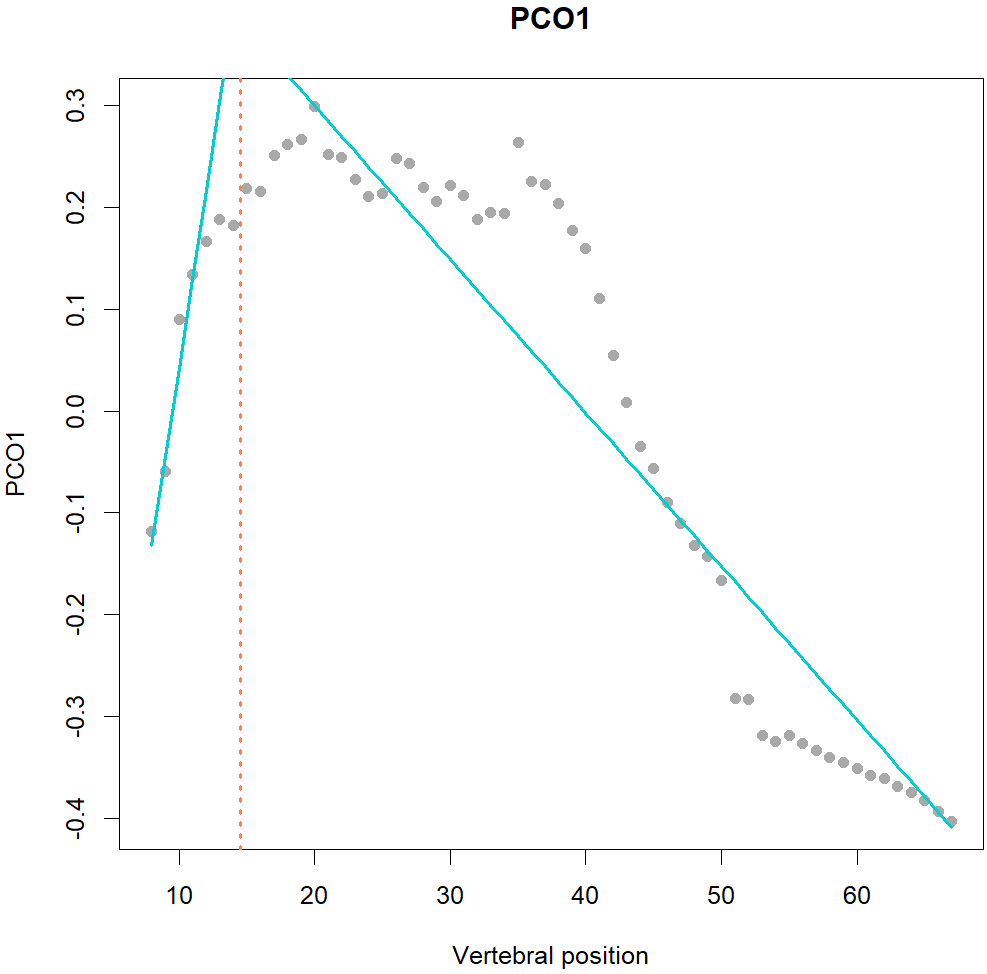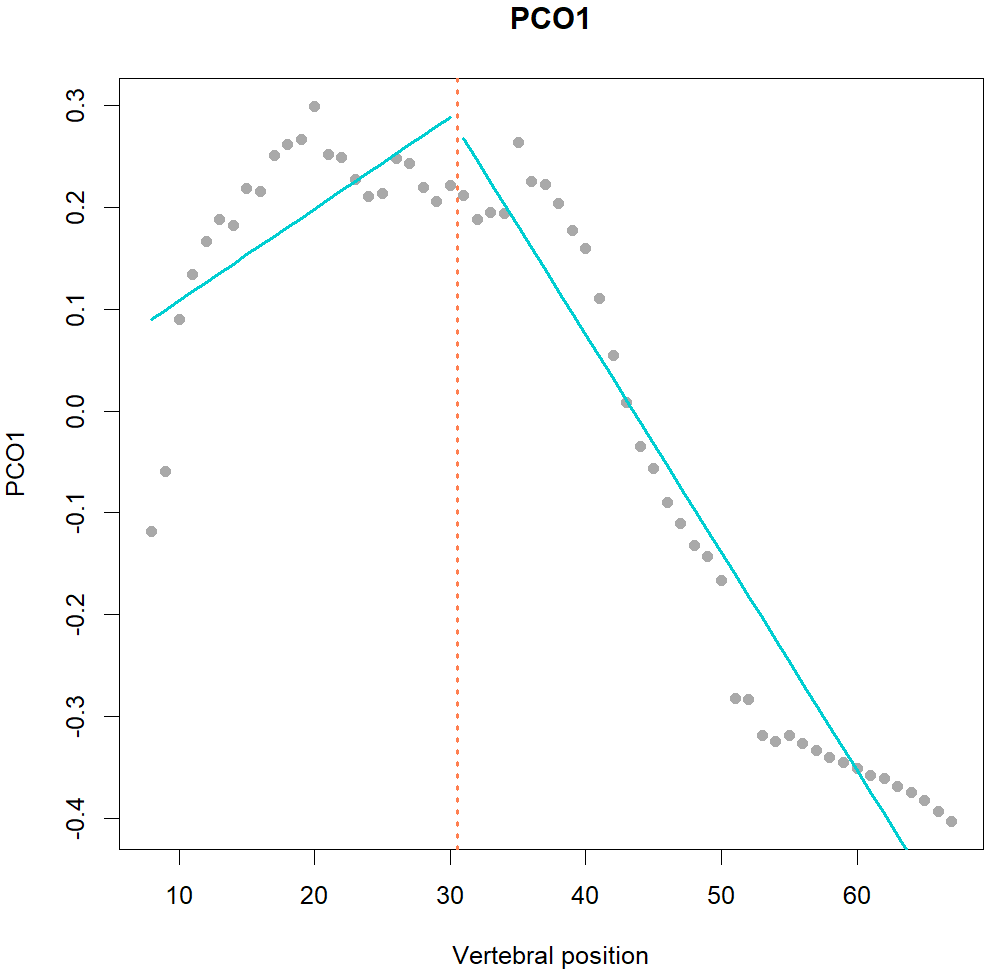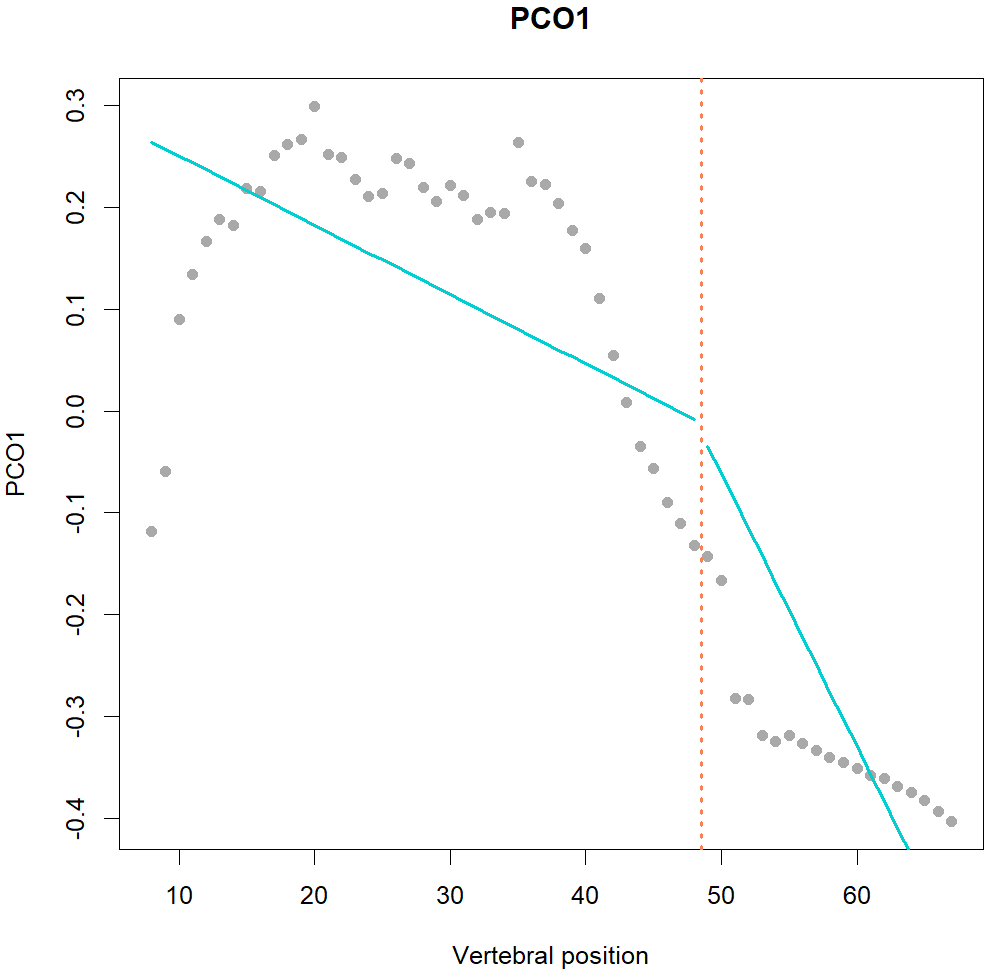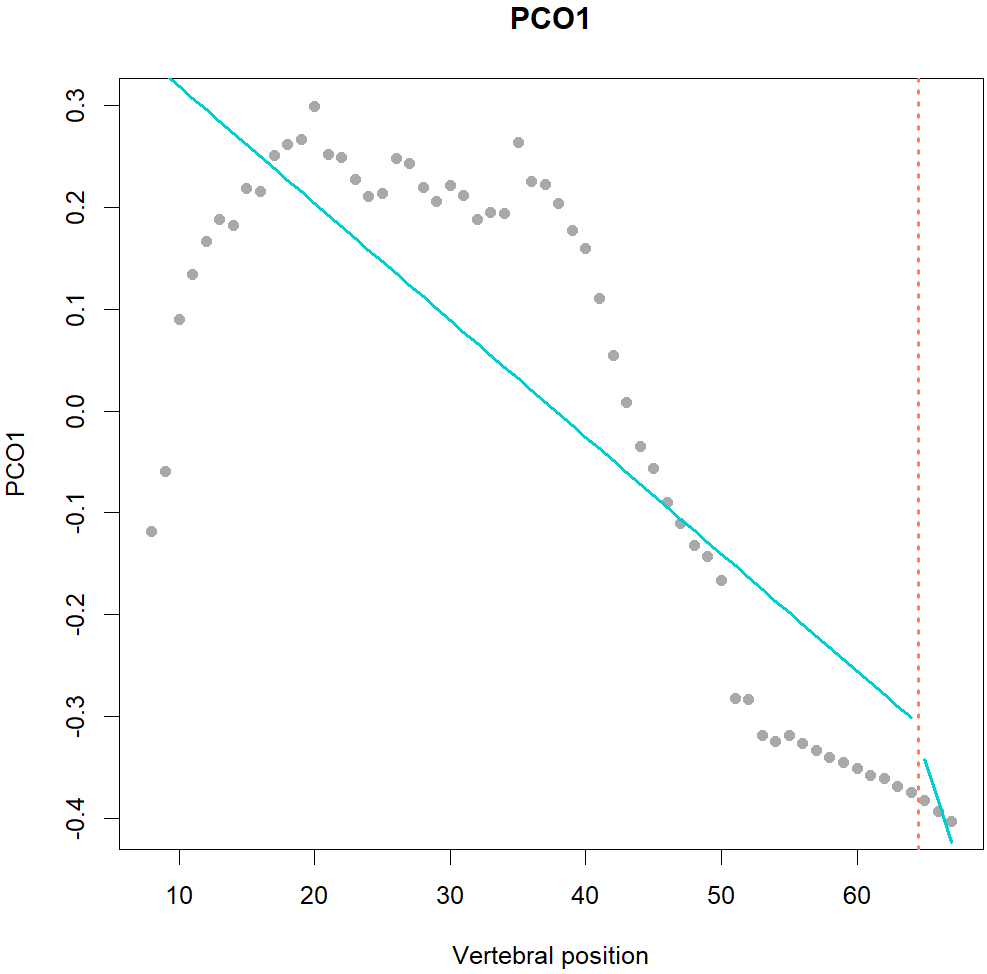
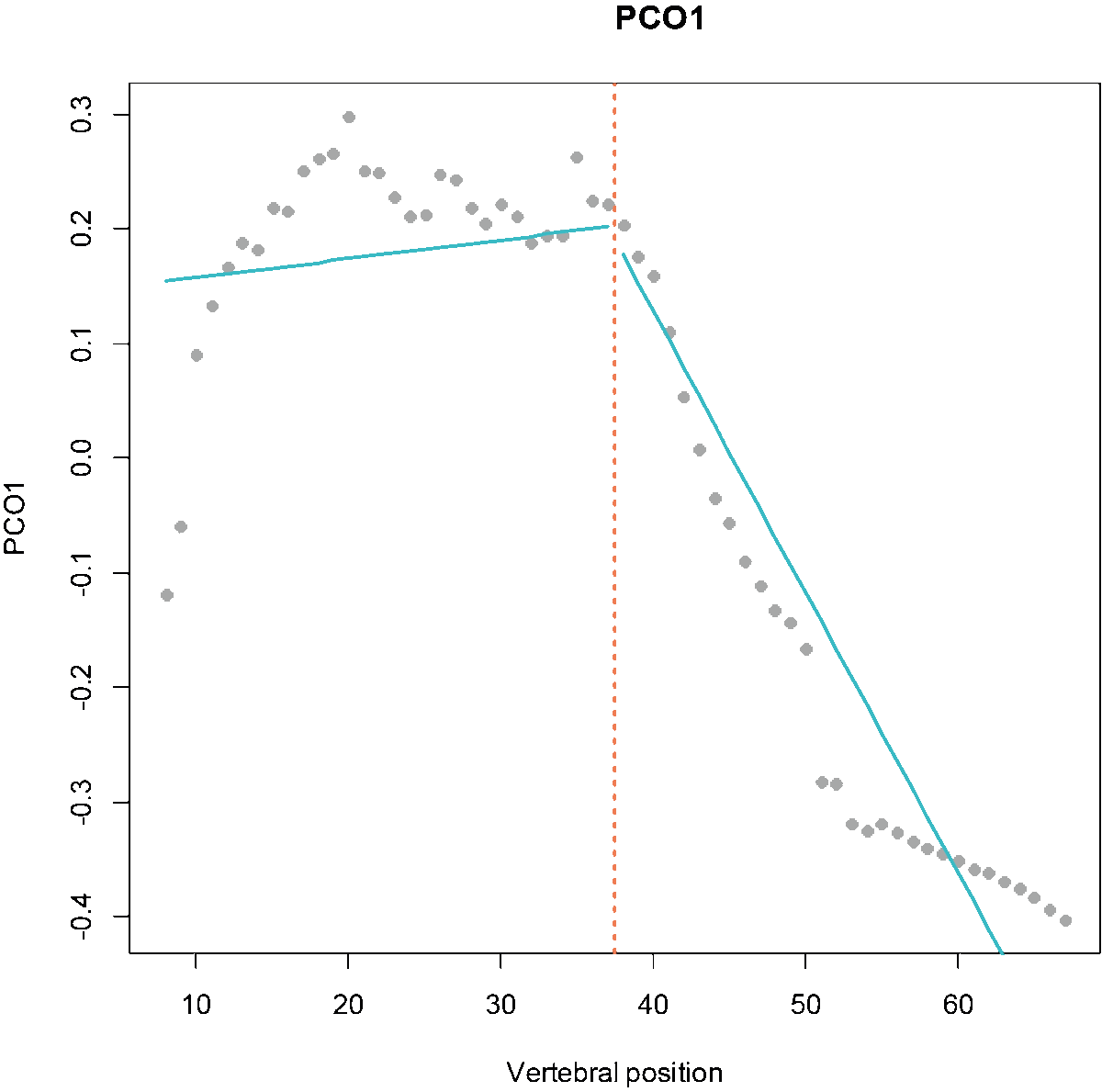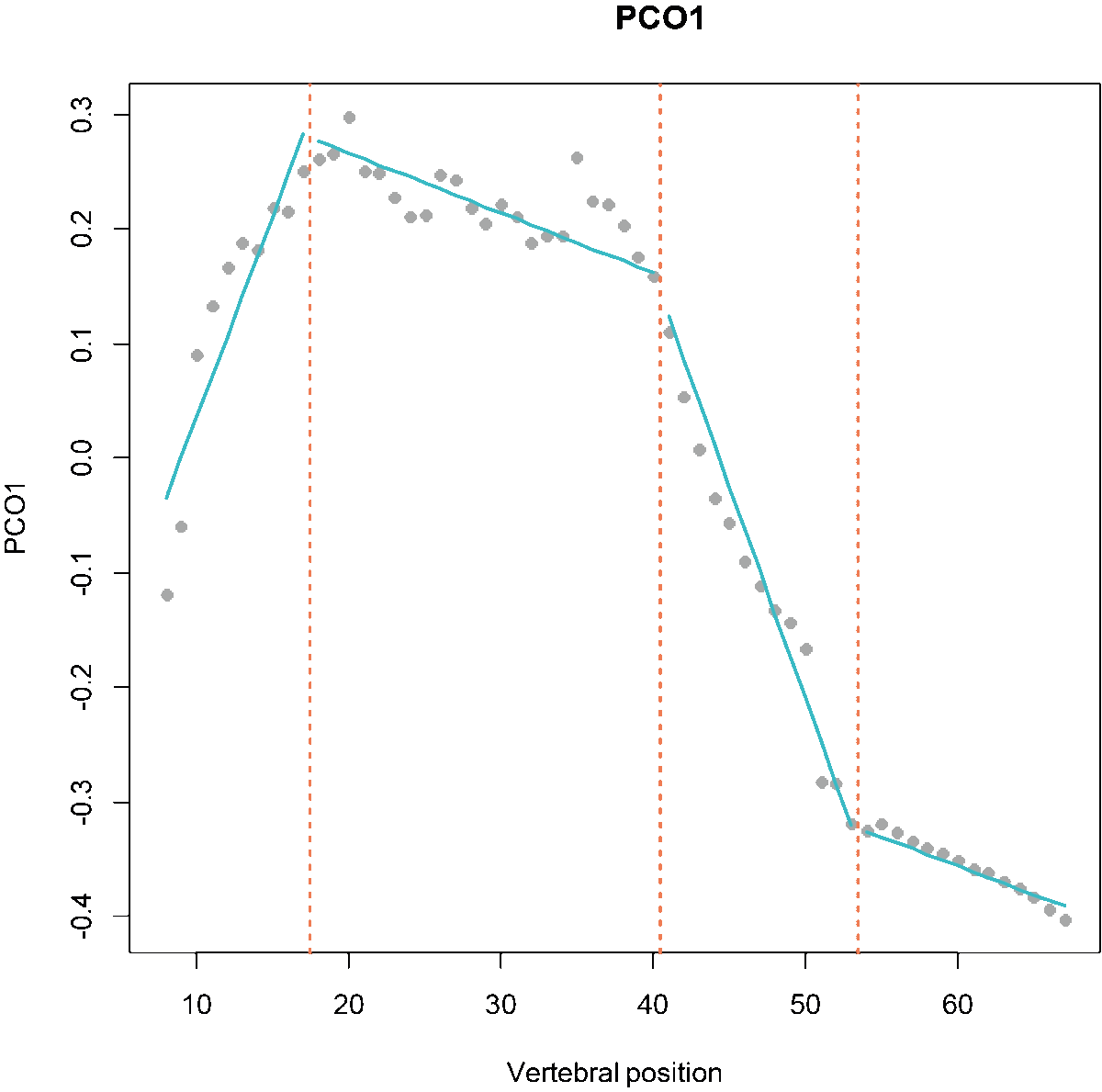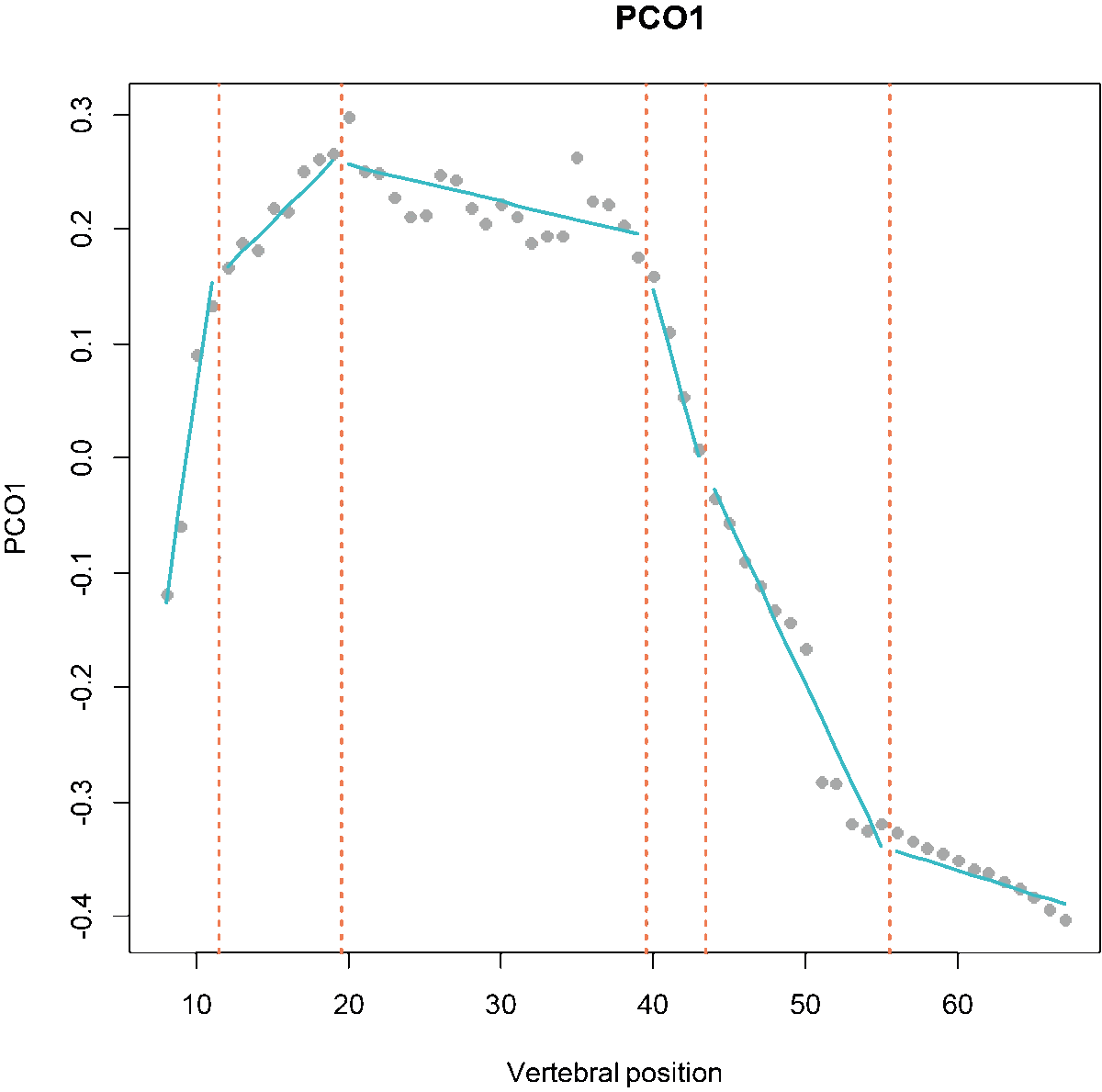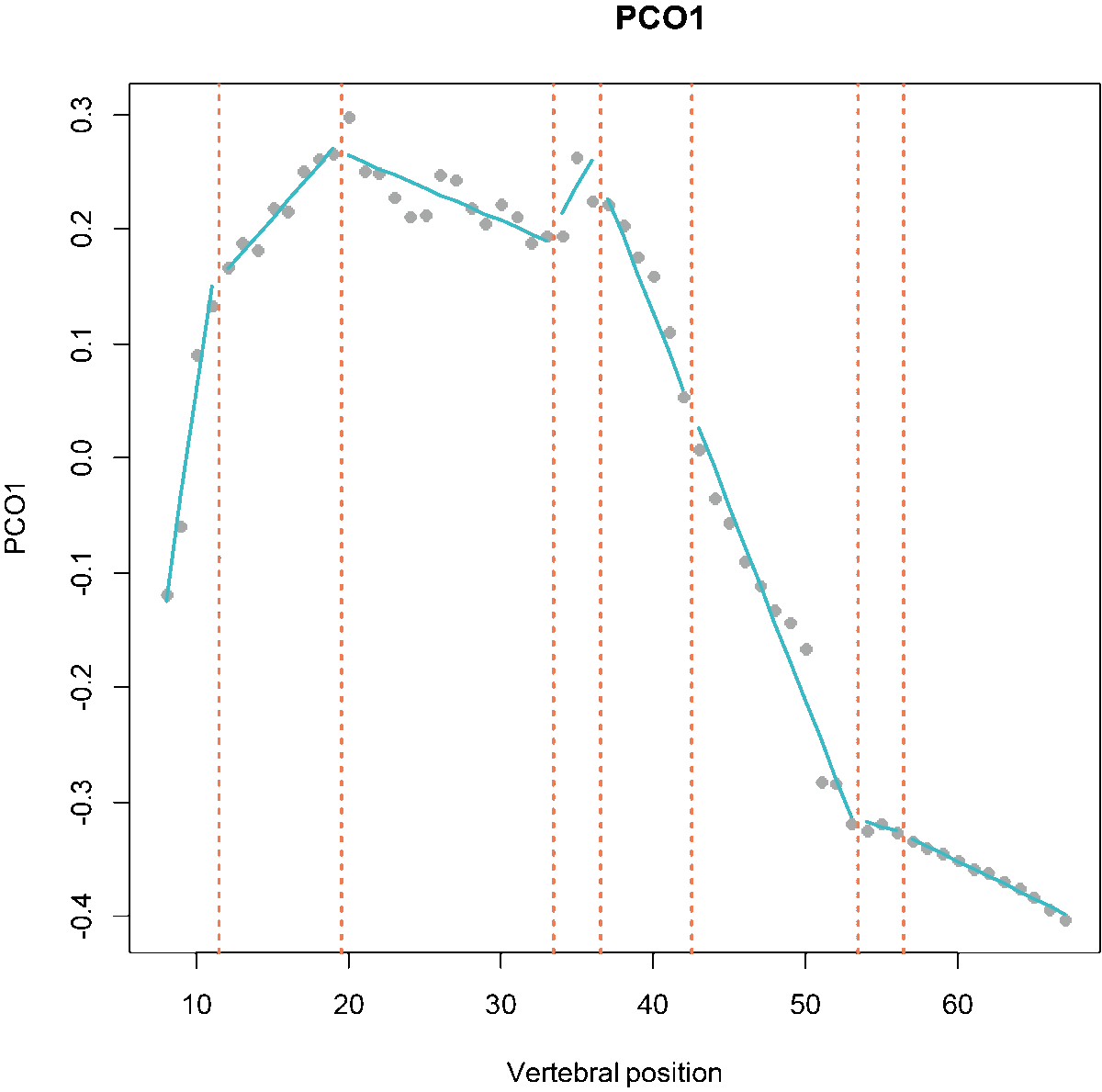
Fig.1. MorphoRegions identifies regions in serially-homologous structures by fitting all possible breakpoint positions for a given number of region and then repeating the method on increasingly complex models.
This package is an updated version of the regions
R-package from Jones et al. (2018) which expanded on previous work
from Head and Polly (2015). This package provides improved
computational methods (reduced time and memory usage) allowing
application to larger datasets. Several features have been added
including the options to perform non-exhaustive searches, run parallel
computing, choose between continuous and discontinuous fitting methods,
define the minimum number of vertebrae in each region, select the best
model using either AICc or BIC, and fit a model to multiple specimens.
Plotting options have also been expanded.
Since version 0.2.0, MorphoRegions can now be applied to both traditional morphometric and geometric morphometric data, providing a unified workflow for identifying regionalization patterns in serially homologous structures.
Package installation
The package is available on CRAN and can be installed and loaded using the following code:
install.packages("MorphoRegions")
library(MorphoRegions)Overview of MorphoRegions worflow
Working with MorphoRegions requires three main steps:
Providing and formatting the data
Performing the regionalization analyses
Visualizing the results
The workflow and functions used to input data into MorphoRegions depend on the type of data as shown in the figure below.
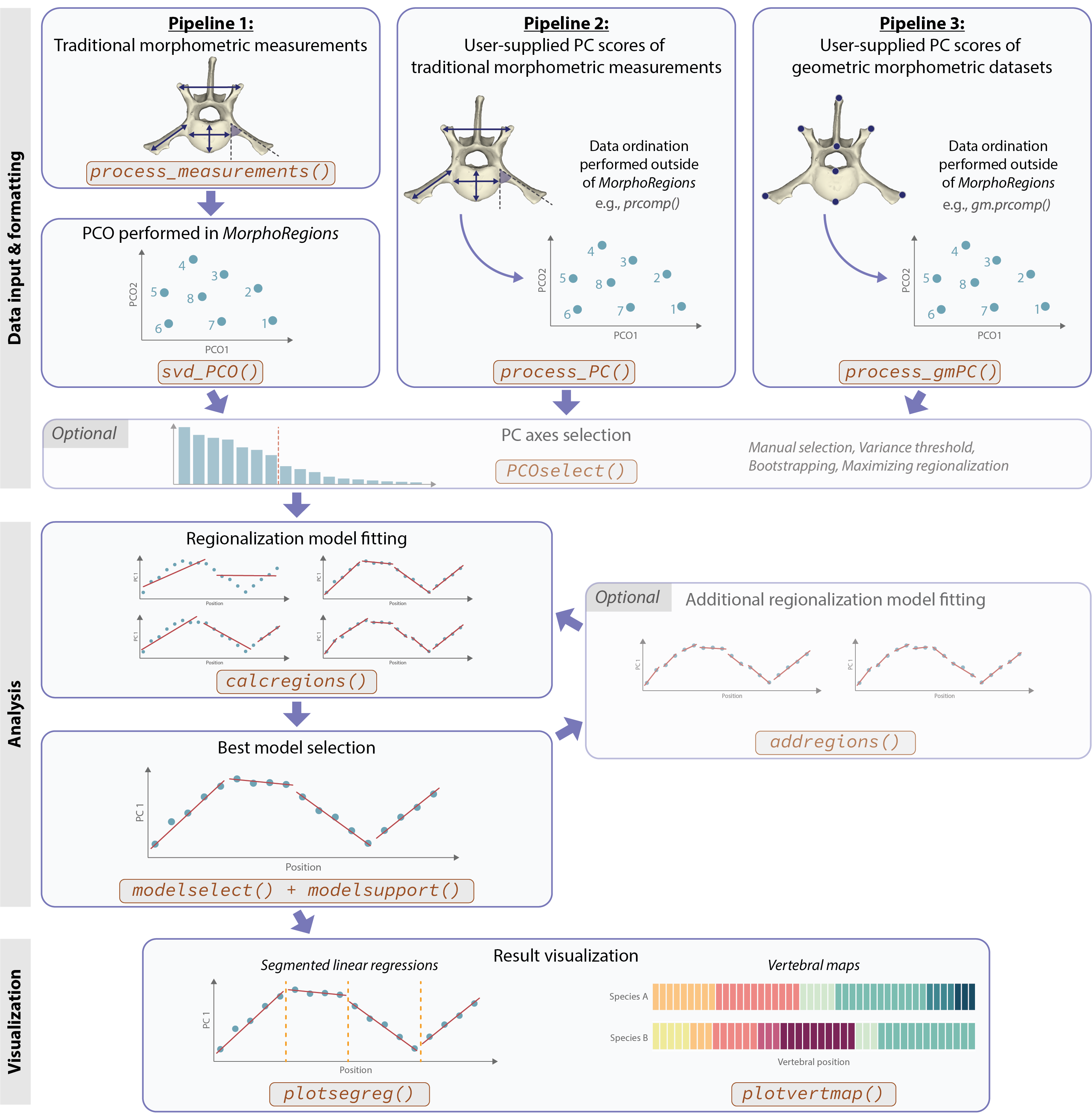
Overview of MorphoRegions workflow and key functions to use. Functions to use for data input depend on the type of data provided.
Preparing the data
MorphoRegions can process the following type of data:
- Raw traditional morphometrics datasets (or other similar dataset where each row is an observation (i.e, serial element) and each column is a variable) (Pipeline 1)
- PC scores (or other type of ordinated data) of traditional morphometric datasets (or similar) (Pipeline 2)
- PC scores of geometric morphometric datasets (Pipeline 3)
The package comes with six built-in datasets.
-
alligator,musm, anddolphincontain measurements on the vertebrae of an alligator, mouse, and dolphin -
porpoisecontains vertebral measurements of three specimens of harbor porpoises and can be used to test fitting a single model for multiple specimens together (see Intraspecific variability) -
seal_gmmcontains Procrustes aligned 3d landmarks on the vertebrae of a seal as well as PC scores -
wolves_gmmcontains Procrustes aligned 3d landmarks and PC scores from 10 subsampled vertebrae along the backbone of two wolves and can be used to test fitting a single model on multiple specimens (see Intraspecific variability)
Dolphin and porpoise data are from Gillet et al. (2022); alligator and mouse data are from Jones et al. (2018); seal 3D landmark data are from Esteban et al. (2023); wolf 3D landmarks are from Schwab et al. (2025).
Raw morphometrics (Pipeline 1)
Data input
Data should be provided as a data frame where each row is an element of the serially-homologous structure (e.g., a vertebra). One column should contain positional information of each element (e.g., vertebral number) and other columns should contain variables that will be used to calculate regions (e.g., morphological measurements).
data("dolphin")
head(dolphin)
#> Vertebra Lc Wc Hc Hnp Wnp Inp Ha Wa Lm Wm Hm Hch Wch Ltp
#> 8 8 1.33 3.37 2.02 2.85 1.17 2.01 1.72 1.48 0.00 0.00 0.0 0 0 1.71
#> 9 9 1.46 3.67 2.10 3.20 1.63 2.01 1.44 1.65 0.00 0.00 0.0 0 0 1.51
#> 10 10 1.57 3.62 2.26 3.13 1.71 2.01 1.42 2.18 0.00 0.00 0.0 0 0 1.06
#> 11 11 1.71 3.75 2.24 3.07 1.71 2.01 1.38 1.25 0.56 0.38 1.7 0 0 1.03
#> 12 12 1.74 3.72 2.28 2.66 1.96 1.99 1.30 1.50 1.45 1.09 2.0 0 0 0.60
#> 13 13 1.82 3.92 2.28 2.61 1.74 1.88 1.29 1.74 1.86 1.12 2.0 0 0 0.37
#> Wtp Itp
#> 8 1.67 1.57
#> 9 1.61 1.57
#> 10 1.90 1.57
#> 11 1.91 1.66
#> 12 1.71 1.57
#> 13 1.44 1.57In the dolphin dataset, the first column contains
positional information (vertebral number) while other columns contain
morphological measurements taken on each vertebra.
Prior to analysis, data must be processed into an object
usable by regions using
process_measurements(). Data should be provided as a
dataframe. The pos argument is used to specify the
name or index of the column containing positional information; if
unspecified, the first column will be used by default. Names or indices
of columns containing variables can be specified with the
measurements argument. The fillNA argument
allows to fill missing values in the dataset. Missing values can be
filled for up to two successive elements (e.g., up to two successive
vertebrae) using a linear imputation (see Jones
et al. (2018) for more details). The
fillNA argument is set to TRUE by default.
dolphin_data <- process_measurements(dolphin, pos = 1)
class(dolphin_data)
#> [1] "regions_data"Data ordination
To allow the analysis of high dimensional datasets, including a
variety of data types, a Principal Coordinates Analysis
(PCO), a distance-based data ordination method, is implemented
in svdPCO(). The function relies on a distance matrix
generated by cluster::daisy(), which tolerates missing
values. Three types of distance metric can be used: Euclidean,
Manhattan, or Gower. Euclidean should only be used where all variables
are similar (e.g., linear measures on the same scale), and is most
similar to a PCA. Gower is good for combining different types of
continuous data (e.g., angles and linear).
dolphin_pco <- svdPCO(dolphin_data, metric = "gower")PCO scores of each vertebra can be plotted along their respective position in backbone (left) or as a morphospace (right).
#Plot PCOs along backbone for PC1 and 2:
plot(dolphin_pco, 1:2)
# Plot morphospace of PC1 and 2:
plot(dolphin_pco, pc_x = 1, pc_y = 2)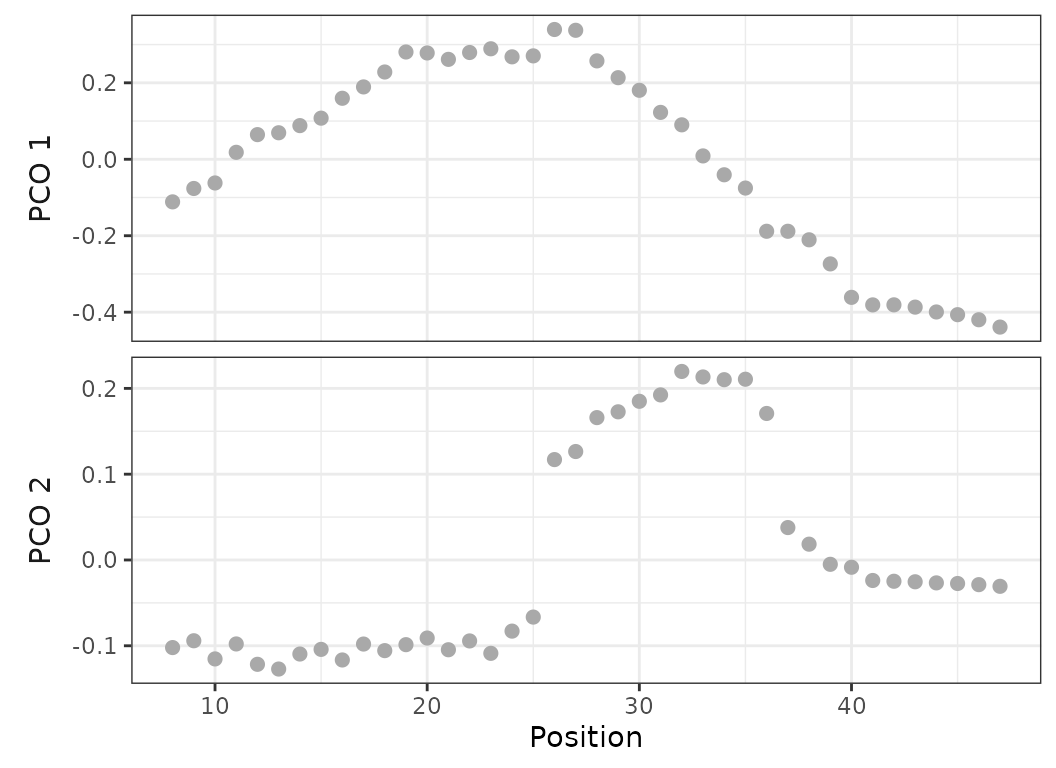
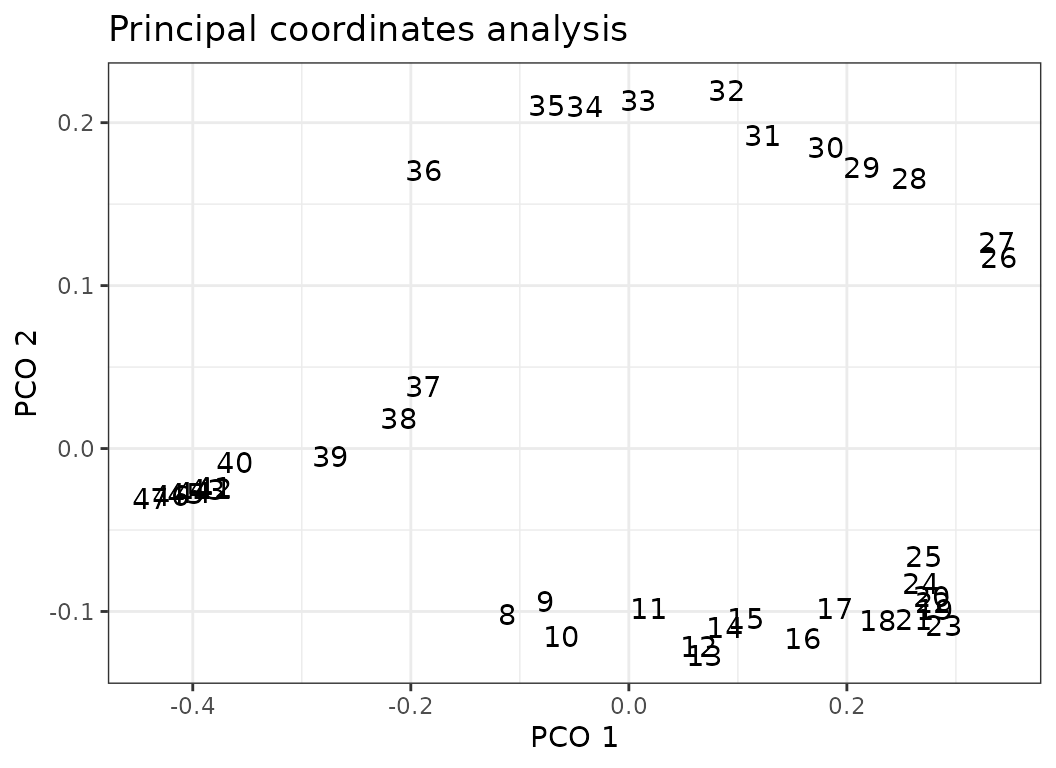
Fig.2. Left: PCO scores of each vertebra along the backbone for PCO1 (top) and PCO2 (bottom). Right: morphospace defined by PCO1 and 2, points plotted as numbers corresponding to the position of each vertebra along the backbone.
PCs from traditional morphometrics (Pipeline 2)
Instead of providing raw data and performing a principal coordinates
ordination, it is possible to directly provide PC scores that have been
computed outside of MorphoRegions, for instance using the
prcomp() function.
# Load dolphin data:
data("dolphin")
# Compute PCA on correlation matrix:
PCA <- prcomp(dolphin[-1], scale=T, center=T)The function process_PC() will then convert the PC
scores into a regions_pco object usable for downstream
analyses. The function requires the PC scores, the eigenvalues, and the
original (raw) data. The positional information of the vertebrae in the
original data and the PC data can be provided with the arguments
pos and posPC, respectively. If these
arguments are not provided, the function will assume that the first
column of the raw data contains the positional information and that the
order of vertebrae in the PC scores data matches the order in the raw
data.
# Extract PC scores and eigenvalues:
PCA_scores <- PCA$x
PCA_eigenval <- PCA$sdev^2
# Input data in MorphoRegions:
dolphin_pca <- process_PC(data = dolphin, pos = 'Vertebra',
pcscores = PCA_scores, eigenvals = PCA_eigenval,
posPC = dolphin[,1])PC scores can then be plotted with the plot() function
as above
PCs from geometric morphometrics (Pipeline 3)
Since v.0.2.0 of the package it also possible to run MorphoRegions on geometric morphometric datasets. As for pipeline 2 above, the data ordination (e.g., PCA) should be performed outside of MorphoRegions.
The seal_gmm dataset is a list containing the 3D
coordinates of 40 Procrustes aligned landmarks from vertebrae 3 to 27 of
a seal (aligned with gpagen from the geomorph
R-package); the PC scores of each vertebrae (obtained using
gm.prcomp() from geomorph), and the eigenvalue
of each PC axis. The positional info of the vertebrae are stored as
dimension names of the landmark coordinates array.
data("seal_gmm")
names(seal_gmm)
#> [1] "coord_gpa" "scores" "eigenvals"
dimnames(seal_gmm$coord_gpa)
#> [[1]]
#> [1] "1" "2" "3" "4" "5" "6" "7" "8" "9" "10" "11" "12" "13" "14" "15"
#> [16] "16" "17" "18" "19" "20" "21" "22" "23" "24" "25" "26" "27" "28" "29" "30"
#> [31] "31" "32" "33" "34" "35" "36" "37" "38" "39" "40"
#>
#> [[2]]
#> [1] "X" "Y" "Z"
#>
#> [[3]]
#> [1] "3" "4" "5" "6" "7" "8" "9" "10" "11" "12" "13" "14" "15" "16" "17"
#> [16] "18" "19" "20" "21" "22" "23" "24" "25" "26" "27"The function process_gmPC() will then convert the PC
scores into a regions_pco object usable for downstream
analyses. The function requires the PC scores, the eigenvalues, and the
landmark coordinates. The positional information of the vertebrae can be
provided as a numeric vector with the pos argument. If no
positional information is provided, the function will extract vertebral
position info form the names of the landmark coordinate array (default)
and convert them to numeric values.
seal_pca <- process_gmPC(data = seal_gmm$coord_gpa,
pcscores = seal_gmm$scores,
eigenvals = seal_gmm$eigenvals)PC scores can then be plotted with the plot() function
as above
Data reduction
While the regionalization analysis can be performed on all PC axes,
it’s likely that lower PC axes mostly contain noise. The number of PC
axes to retain for analysis can be selected using
PCOselect().
Four different methods to select PCs are available
and be specified using the method argument:
-
"manual": the number of PCs is directly defined by the user -
"variance": PC axes with a variance above a given threshold are retained -
"max": computes the minimum number of PCs axes needed to maximize the number of regions -
"boot": PCO axes are selected through bootstrapping by comparing observed eigenvalues to eigenvalues from randomized data and retaining PCO axes with eigenvalues higher than values from randomized data. When using this method, the screeplot showing observed and simulated eigenvalues can be plotted. Note that bootstrapping is sensitive to unequal variances of variables and should only be used on data that have been scaled (settingscale = TRUEin the original call tosvdPCO()).
Manual selection:
The number of PCs is directly defined via the scores
argument. For instance, selecting PC axes 1 to 5:
PCOs <- PCOselect(dolphin_pco, method = "manual",
scores = 5)
PCOs
#> A `regions_pco_select` object
#> - PC scores selected: 1, 2, 3, 4, 5
#> - Method: manualVariance cutoff:
PC axes with a variance (defined as the ratio between the eigenvalue
of the given axis and the sum of all eigenvalues) higher to the
cutoff value (expressed as a value between 0 and 1) are
retained. For instance, selecting PC axes with variance above 5%:
PCOs <- PCOselect(dolphin_pca, method = "variance",
cutoff = .05)
PCOs
#> A `regions_pco_select` object
#> - PC scores selected: 1, 2, 3
#> - Method: variance (cutoff: 0.05)Maximizing the number of regions:
PCs axes needed to obtain the maximum number of regions will be
retained. The segmented models (output from calcregions)
must be supplied to the results argument. The criterion
(AICc or BIC) used to maximize the region score should be defined with
criterion (see the section Regionalization
analysis below for details on running calcregions and
criterion selection) For instance, maximizing the number of PC axes to
retain for the seal geometric morphometric dataset:
# Max:
regionresults <- calcregions(seal_pca, scores = 1:24, noregions = 4,
minvert = 3, cont = TRUE, exhaus = TRUE)
PCOs <- PCOselect(seal_pca, method = "max",
results = regionresults,
criterion = "bic")
PCOs
#> A `regions_pco_select` object
#> - PC scores selected: 1
#> - Method: max (criterion: BIC)
plot(PCOs)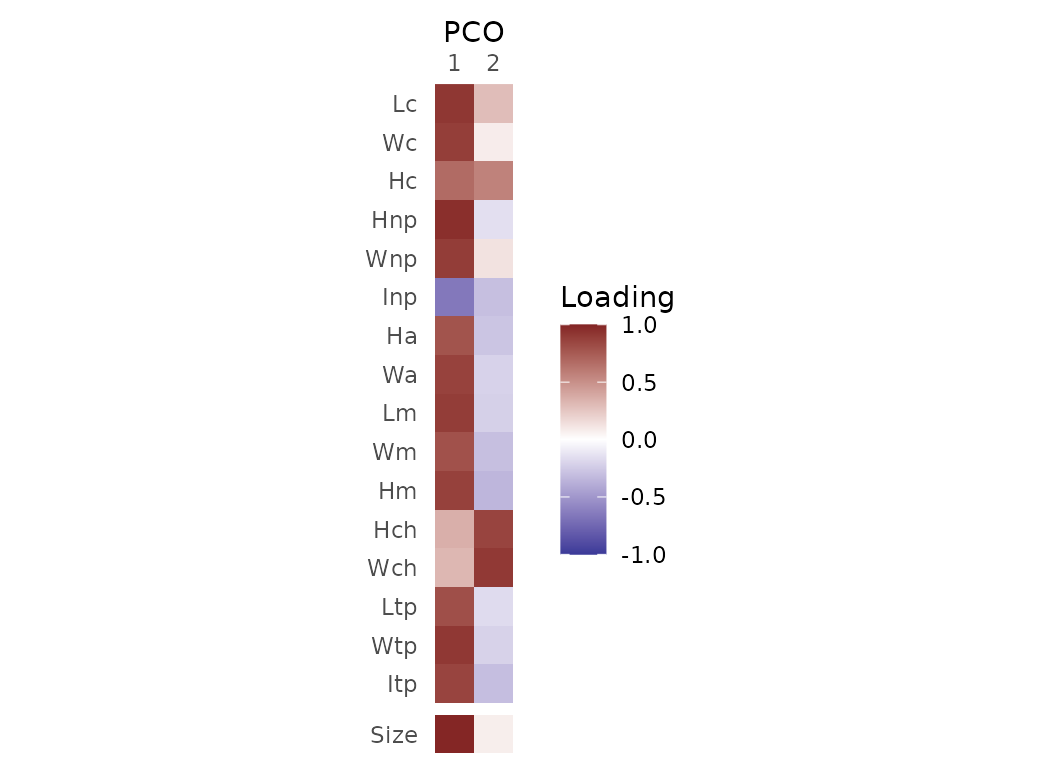
Fig.3a. Graph of region scores (left y-axis) and cumulative variance (right y-axis) obtained with varying number of PCO axes included for AICc (left graph) and BIC (right graph). Blue triangles correspond to region score obtained for each individual PCO axis and orange circle to region scores obtained with cumulative number of PCO axes. The cumulated variance explained by PCO axes is represented by the grey curve.
Bootstrap:
PC axes are selected through bootstrapping by comparing observed
eigenvalues to eigenvalues from randomized data. PC axes with
eigenvalues higher than values from randomized data are retained. When
using this method, the screeplot showing observed and simulated
eigenvalues can be plotted. Because the function needs to recompute the
ordination based on randomized data, this option is only
available for PCO axes computed within MorphoRegions
(pipeline 1). Note that bootstrapping is sensitive to unequal
variances of variables and should only be used on data that have been
scaled (setting scale = TRUE in the original call to
svdPCO()).
# Bootstrap:
PCOs <- PCOselect(dolphin_pco, method = "boot")
PCOs
#> A `regions_pco_select` object
#> - PC scores selected: 1, 2
#> - Method: boot (500 replications)
plot(PCOs)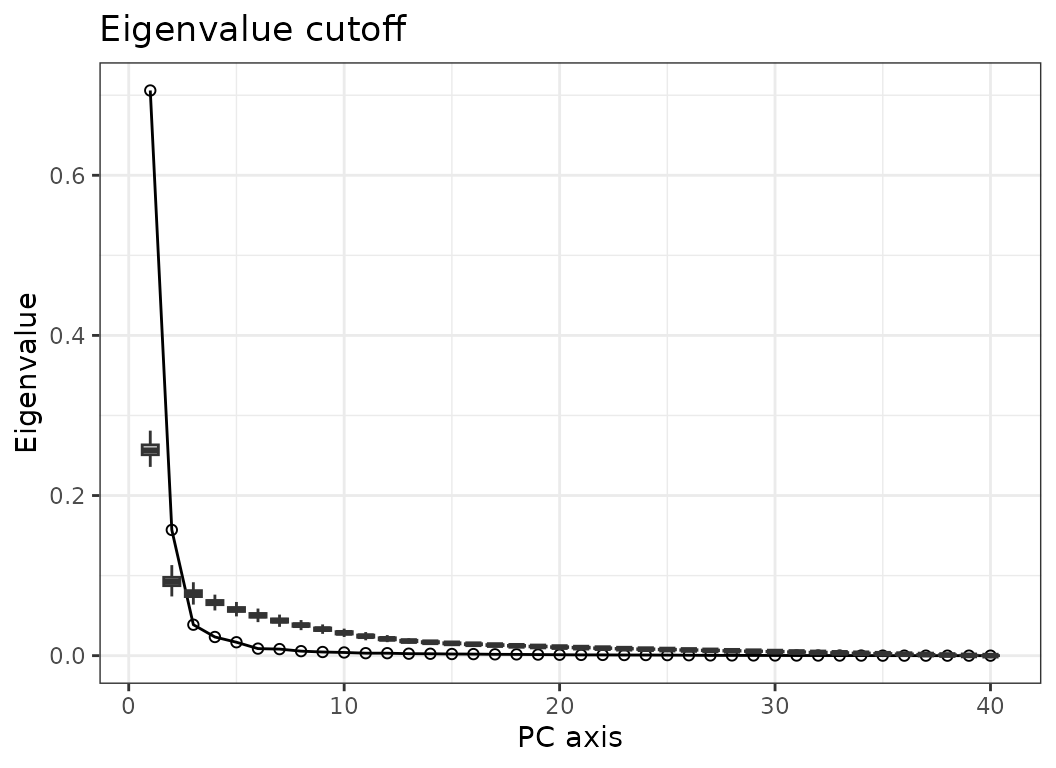
Fig.3b. Screeplot of observed eigenvalues (line) and eigenvalues from randomized data (boxes). Here PCO axes 1 and 2 are retained for subsequent analyses.
Variable loadings
Correlations between original variables and each PCO axis are
obtained using PCOload(). The additional ‘Size’ variable
corresponds to the mean value of the variables. Note that This
function is not available for geometric morphometric datasets (pipeline
3).
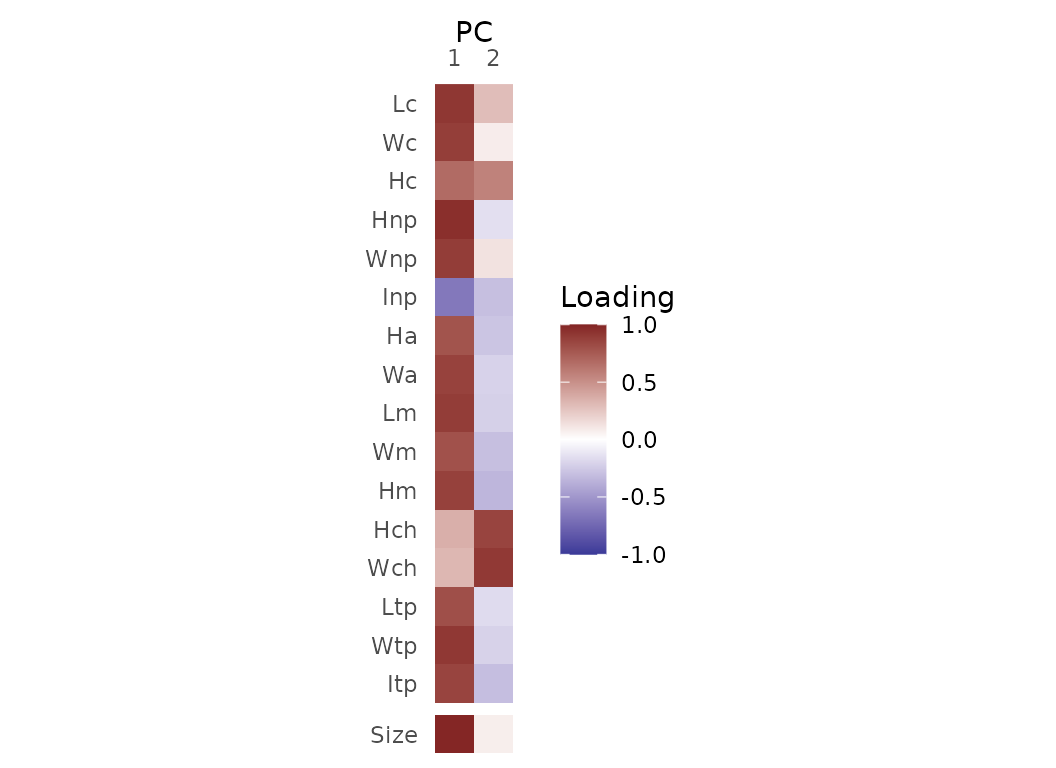
Fig.4. Correlations between original variables and PCO axes 1 and 2.
Regionalization analysis
The regionalization analysis is performed in three steps:
Fitting all possible models for all number of regions requested
Selecting the best model for each number of regions tested
Selecting the overall best model
Fitting segmented linear regressions
calcregions() fits all possible combinations of
breakpoints (boundaries between successive regions) for each number of
regions to be tested. The analysis will fit models on PCO scores, so the
output of svdPCO() needs to be provided. The number of PCOs
to keep (supplied either as a vector or as the output of
PCOselect()), and the maximum number of regions to test
need to be provided with the scores and
noregions arguments, respectively.
The function also offers several optional arguments (see the Model fitting options section for details):
-
minvert: define the minimum number of vertebrae (elements) in each region (see here) -
cont: choose to fit either a continuous fit (TRUE) or discontinuous fit (FALSE) (see here) -
includebp: force breakpoint(s) to fall at the supplied value(s) (see here) -
omitbp: exclude breakpoint(s) falling at the supplied value(s) (see here) -
exhaus: either to fit all possible combinations (TRUE) or only the most probable ones (FALSE) to reduce computing time (see here) -
cl: option to use parallel computation (see here)
regionresults <- calcregions(dolphin_pco, scores = PCOs, noregions = 5,
minvert = 3, cont = TRUE,
exhaus = TRUE, cl = NULL)The output of the function contains information on the parameters
used to run the analysis and total number of models tested while using
summary() allows one to see how many models are possible
and how many have been tested for each given number of regions.
regionresults
#> A `regions_results` object
#> - number of PCOs used: 2
#> - number of regions: 1, 2, 3, 4, 5
#> - model type: continuous
#> - min vertebrae per region: 3
#> - total models saved: 28810
#> Use `summary()` to examine summaries of the fitting process.
summary(regionresults)
#> Regions Possible Tested Saved Comp. method Saving method
#> 1 1 1 1 Exhaustive All
#> 2 35 35 35 Exhaustive All
#> 3 528 528 528 Exhaustive All
#> 4 4495 4495 4495 Exhaustive All
#> 5 23751 23751 23751 Exhaustive AllSelecting best model for each number of regions
modelselect() retains the best model for each number of
regions tested. The best models are selected as the ones with the lowest
residual sum of squares (RSS).
models <- modelselect(regionresults)
models
#> Regions BP 1 BP 2 BP 3 BP 4 sumRSS RSS.1 RSS.2
#> 1 . . . . 1.898 1.456 0.441
#> 2 26 . . . 0.413 0.105 0.308
#> 3 23 29 . . 0.147 0.092 0.055
#> 4 23 30 40 . 0.073 0.034 0.040
#> 5 23 27 34 40 0.046 0.026 0.020Each row of the output corresponds to the best model for a given
number of regions. Columns named BP give the position of
region boundaries (or breakpoints), each value corresponding to the
position of the last element (vertebra) of a given region. The other
columns provide the total residual sum of squares across all PC axes
(sumRSS) and the residual sum of square for each PC axis
individually (RSS.).
In the example above, in the model with three regions, the first region encompasses vertebrae 8 (first vertebra in the dataset) to 23, the second contains vertebrae 24 to 29, and the last region contains vertebrae 30 to 47 (last vertebra in the dataset).
Selecting best overall model and region score
modelsupport() will find the best overall models across
the models returned by modelselect(). The best model is
calculated using AICc or BIC which allows the selection
of the best fit model while penalizing for models that are too complex.
See Jones et al. (2018) for AICc calculations and Gillet et al. (2024) for BIC calculations.
supp <- modelsupport(models)
supp
#> - Model support (AICc)
#> Regions BP 1 BP 2 BP 3 BP 4 sumRSS AICc deltaAIC model_lik Ak_weight
#> 5 23 27 34 40 0.046 -556.036 0.000 1 1
#> 4 23 30 40 . 0.073 -528.096 27.940 0 0
#> 3 23 29 . . 0.147 -480.952 75.084 0 0
#> 2 26 . . . 0.413 -405.787 150.250 0 0
#> 1 . . . . 1.898 -290.769 265.267 0 0
#> Region score: 5
#>
#> - Model support (BIC)
#> Regions BP 1 BP 2 BP 3 BP 4 sumRSS BIC deltaBIC model_lik BIC_weight
#> 5 23 27 34 40 0.046 -526.559 0.000 1 1
#> 4 23 30 40 . 0.073 -502.645 23.914 0 0
#> 3 23 29 . . 0.147 -460.321 66.238 0 0
#> 2 26 . . . 0.413 -390.668 135.891 0 0
#> 1 . . . . 1.898 -281.774 244.784 0 0
#> Region score: 5Models are ordered from best to worst fit. Here, both AICc and BIC find the model with 5 regions as the best overall model with the following regions: region1: vertebrae 8 to 23, region2: vertebrae 24 to 27, region3: 28 to 34, region4: 35 to 40, region5: 41 to 47.
In addition to returning the best models, the function also calculates the region score, a continuous value accounting for the fact models with different number of regions could fit the data well. The region score is calculated as the sum of the number of region in each model weighted by their respective AICc/BIC weight. In this example, the region score for BIC is calculated as .
Adding more regions
In the example above, the model with 5 regions is by far the best one
(AICc/BIC weight = 1). In such cases where the most complex model is the
best model, it’s likely that more complex models fit the data better.
addregions() allows one to fit additional models
without the need to recompute models with fewer regions.
regionresults <- addregions(regionresults, noregions = 6:7, exhaus = TRUE)
summary(regionresults)
#> Regions Possible Tested Saved Comp. method Saving method
#> 1 1 1 1 Exhaustive All
#> 2 35 35 35 Exhaustive All
#> 3 528 528 528 Exhaustive All
#> 4 4495 4495 4495 Exhaustive All
#> 5 23751 23751 23751 Exhaustive All
#> 6 80730 80730 80730 Exhaustive All
#> 7 177100 177100 177100 Exhaustive AllIdentifying the best model now that models with 6 and 7 regions have been added:
models <- modelselect(regionresults)
supp <- modelsupport(models)
supp
#> - Model support (AICc)
#> Regions BP 1 BP 2 BP 3 BP 4 BP 5 BP 6 sumRSS AICc deltaAIC model_lik
#> 7 19 24 27 35 38 41 0.021 -596.668 0.000 1.000
#> 6 19 24 27 34 40 . 0.026 -590.493 6.174 0.046
#> 5 23 27 34 40 . . 0.046 -556.036 40.632 0.000
#> 4 23 30 40 . . . 0.073 -528.096 68.572 0.000
#> 3 23 29 . . . . 0.147 -480.952 115.715 0.000
#> 2 26 . . . . . 0.413 -405.787 190.881 0.000
#> 1 . . . . . . 1.898 -290.769 305.899 0.000
#> Ak_weight
#> 0.956
#> 0.044
#> 0.000
#> 0.000
#> 0.000
#> 0.000
#> 0.000
#> Region score: 6.96
#>
#> - Model support (BIC)
#> Regions BP 1 BP 2 BP 3 BP 4 BP 5 BP 6 sumRSS BIC deltaBIC model_lik
#> 7 19 24 27 35 38 41 0.021 -562.017 0.000 1.000
#> 6 19 24 27 34 40 . 0.026 -557.902 4.116 0.128
#> 5 23 27 34 40 . . 0.046 -526.559 35.459 0.000
#> 4 23 30 40 . . . 0.073 -502.645 59.373 0.000
#> 3 23 29 . . . . 0.147 -460.321 101.697 0.000
#> 2 26 . . . . . 0.413 -390.668 171.349 0.000
#> 1 . . . . . . 1.898 -281.774 280.243 0.000
#> BIC_weight
#> 0.887
#> 0.113
#> 0.000
#> 0.000
#> 0.000
#> 0.000
#> 0.000
#> Region score: 6.89The best overall model now is the model with 7 regions both when using AICc and BIC. Since the best model is still the most complex model, additional model(s) with more than seven regions should be fitted (not run here for computational reasons).
Model performance
modelperf() returns the univariate (for each PC axis
individually) and multivariate (all PC axes)
and adjusted
for a given model. The
can be calculated for the best model (model = 1) for a
given criterion (criterion argument), but also the second
best (model = 2), third best (model = 3), etc.
In this case, the output of modelsupport() must be
supplied.
modelperf(dolphin_pco, scores = PCOs, modelsupport = supp,
criterion = "bic", model = 1)
#> Breakpoints: 19, 24, 27, 35, 38, 41
#>
#> - Univariate:
#> R² Adj. R²
#> PCO.1 0.996 0.995
#> PCO.2 0.982 0.978
#>
#> - Multivariate:
#> R² Adj. R²
#> 0.993 0.992Alternatively, it is also possible to calculate the
for a user-defined model using the bps argument specifying
the position of breakpoints (region boundaries) of the model to
test.
Breakpoint position variability
calcBPvar() calculates the variability in breakpoint
position for a given number of regions (noregions argument)
using weighted means and standard deviation based either on AICc or BIC
weight (defined by criterion argument). The variability can
be calculated on a given proportion of best models across all fitted
models from this number of regions (pct argument).
Here is an example of calculating the variability of breakpoint positions for models with 7 regions, using the top 10% best models evaluated with the BIC criterion.
bpvar <- calcBPvar(regionresults, noregions = 7,
pct = 0.1, criterion = "bic")
bpvar
#> BP 1 BP 2 BP 3 BP 4 BP 5 BP 6
#> wMean 19.214 23.973 26.983 34.911 37.965 41.017
#> wSD 0.693 0.347 0.227 0.645 0.349 0.238
#> - Computed using top 10% of modelsNote that when using output from calcBPvar for plotting,
breakpoint positions will be rounded to the closest whole number. In the
example above, breakpoints will then be 19, 24, 26, 35, 38, and 41.
Since calcregions only returns whole numbers as breakpoint
values, a weighted mean breakpoint position of 23.97 involves that most
models found a breakpoint value of 24 (corresponding to a region break
between vertebrae 24 and 25).
Visualizing results
MorphoRegions offers two main plotting options to visualize the results: 1) scatter plots showing PC scores of each element (vertebra) along the structure (backbone) and the segmented linear regressions, 2) vertebral maps showing the regional identity of each element (vertebra).
Segmented linear regression plots
The scatter plots of the segmented linear regressions can be plotted
using plotsegreg(). The output of svdPCO() and
the number of the PC axis to plot (scores argument) must be
supplied.
The function can either be used to plot the best (or second best,
third best, etc.) model (model argument) for a given
criterion, by supplying the output of modelsupport().
Alternatively, a specific model can also be plotted by providing the
position of the breakpoints (bps argument).
# Plot regressions for best model:
plotsegreg(dolphin_pco, scores = 1:2,
modelsupport = supp,
criterion = "bic",
model = 1)
# Plot regressions for a specific model:
plotsegreg(dolphin_pco, scores = 1,
bps = c(15, 24, 37),
cont = TRUE)

Fig.5. Scatterplots showing PCO scores and segmented regressions on PC1 and PC2 for the best model (left) and on PC1 for a 4 regions model with breakpoints at vertebrae 15, 24, and 37 (right).
Vertebral maps
plotvertmap() produces vertebral maps, which are plots
in which each rectangle corresponds to an element (i.e., vertebra) that
is color-coded according to the region to which it belongs. The function
provides many different plotting options.
The function requires regions_data (from
process_measurements()), regions_pco (from
svdPCO()), or regions_sims (from
simregions(); see below) to be provided. The positional
information of each element (i.e., vertebral number) will be extracted
from one of these objects.
The regional identity of each element can be defined either by
providing a regions_modelsupport object (from
modelsupport()) or by directly specifying the position of
breakpoints of a model.
#Plot vertebral map using best BPs from modelsupport (BIC)
plotvertmap(dolphin_pco, name = "Dolphin A",
modelsupport = supp, criterion = "bic", model = 1)
#Plot vertebral map using 2nd best BPs from modelsupport (AICc)
plotvertmap(dolphin_pco, name = "Dolphin B",
modelsupport = supp, criterion = "aic", model = 2)
#Plot vertebral map using arbitrary breakpoints
plotvertmap(dolphin_pco, name = "Dolphin C",
bps = c(12, 17, 20, 30))


Fig.6. Vertebral maps of the best fit model using BIC criterion (top), the 2nd best fit model using AICc criterion (middle), a user-defined model with breakpoints at vertebrae 12, 17, 20, and 30 (bottom).
Here the seven first elements (rectangles corresponding each to one
vertebra) are grayed as they were not included in the analyses. It is
possible to hide elements excluded from the analyses by setting the
dropNA argument to TRUE.
Variability in breakpoint positions between regions
can also be added either by supplying a regions_BPvar
object (from calcBPvar()) or a user-defined vector. When a
regions_BPvar object is supplied, the position of
breakpoints corresponds to the weighted mean position computed by
calcBPvar().
# BP variability from calcBPvar:
plotvertmap(dolphin_pco, name = "Dolphin",
bpvar = bpvar, dropNA = TRUE)
# BP variability from user-defined vector, plotting SD bars in grey:
plotvertmap(dolphin_pco, name = "Dolphin",
modelsupport = supp,
criterion = "bic",
bp.sd = c(2.2, 1.4, 2.5, 1.5, 2.1, 0.8),
sd.col="#636363", dropNA = TRUE)

Fig.7. Vertebral maps showing breakpoint position variability calculated from calcBPvar (top) and with values directly supplied to the function (bottom). Dots correspond to breakpoint position and horizontal lines correspond to the standard deviation of each breakpoint position.
To facilitate the interpretation of the regions, the function offers some plotting options.
It is possible to add other boundaries on the plot,
such as the boundaries between traditional anatomical regions
(thoraco-lumbar, lumbo-caudal, etc). Boundary positions can be supplied
as a vector to the reg.lim argument, with each numeric
value corresponding to the position of the last element before the
boundary. For example, in the dolphin dataset, the thoraco-lumbar,
lumbo-caudal, and caudal-fluke transitions fall between vertebrae 17-18,
25-26, and 35-36, respectively.
It is also possible to plot the positional info of each element (vertebra) as a number on the corresponding rectangle.
# Remove missing vertebrae, add vertebrae number
# and add thoraco-lumbar, lumbo-caudal, and caudal-fluke boundaries:
plotvertmap(dolphin_pco, name = "Dolphin", modelsupport = supp,
criterion = "bic", dropNA = TRUE,
reg.lim = c(17 ,25, 35),
lim.col = c("red", "blue", "green"))
# Add positional info of each vertebra:
plotvertmap(dolphin_pco, name = "Dolphin", modelsupport = supp,
criterion = "bic", dropNA = TRUE, text = TRUE)

Fig.8. Vertebral maps with anatomical region boundaries represented by vertical bars (top) and vertebral position labelled on corresponding rectangle (bottom).
By default, the function plots the elements (vertebrae) as count
(vertebral count) but it is also possible to plot elements as
percentage of the total number of elements (by setting
type = "percent").
In both cases, each element on the plot will have the same size.
Alternatively, it is possible to scale rectangles on the plot
according to the relative size of each element by supplying a
vector containing size information to the centraL argument.
In the example below, rectangles are scaled according to the centrum
length of each vertebra (supplied in the Lc column of the
original dataset).
# Plot vertebrae as percentage of total count:
plotvertmap(dolphin_pco, name = "Dolphin",
type = "percent",
modelsupport = supp, criterion = "bic",
dropNA = TRUE, reg.lim = c(17, 25, 35))
# Scale vertebrae relative to their centrum length (scale in cm):
plotvertmap(dolphin_pco, name = "Dolphin",
type = "count", centraL = "Lc",
modelsupport = supp, criterion = "bic",
dropNA = TRUE, reg.lim = c(17, 25, 35))
# Scale vertebrae relative to their centrum length (scale in % total length):
plotvertmap(dolphin_pco, name = "Dolphin",
type = "percent", centraL = "Lc",
modelsupport = supp, criterion = "bic",
dropNA = TRUE, reg.lim = c(17, 25, 35),
lim.col = "grey")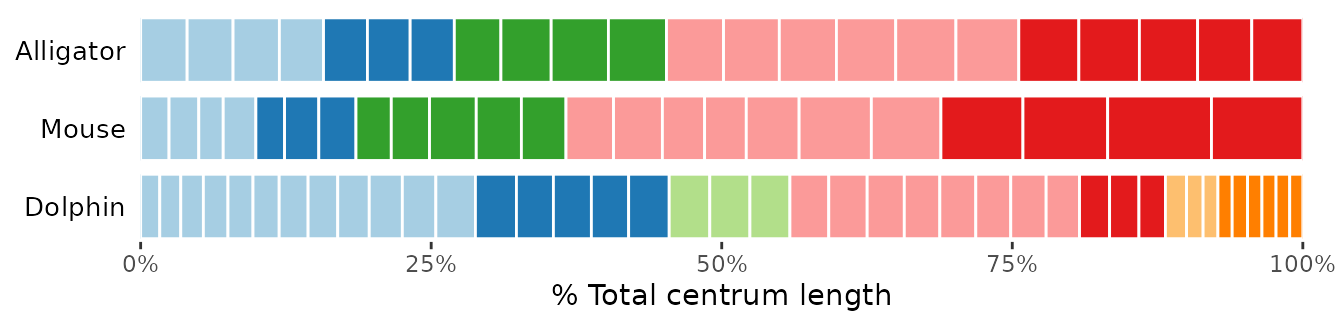


Fig.9. Vertebral maps plotting vertebrae as percentage of total vertebral count (top), scaled to their centrum length relative to total length in absolute value (middle), and scaled to their centrum length in percentage (bottom).
Finally, it is possible to change colors used for each
region with the block.cols argument. Colors can
either be supplied as a vector, in which case regions will be
color-coded following the order of colors supplied in the
vector.
Alternatively, it’s also possible to define a set of colors per
block, each block being defined by the block.lim
argument. In this case, colors must be supplied as a named list, each
element of the list containing a vector of color for a given block. For
instance, for the dolphin data, we could define regions in the precaudal
block (thoracic + lumbar) to be colors in shades of beige/brown, while
regions in the caudal block (caudal + fluke) to be color in shades of
blue. Since the lumbo-caudal transition falls between vertebrae 25 and
26, we’ll define two blocks with a limit at vertebra 25.
# Manually color-coding each region using the viridis palette:
library(viridisLite)
nreg <- supp$Model_support_BIC[1, 1] # get nbr of regions
cols <- viridis(n = nreg)
plotvertmap(dolphin_pco, name = "Dolphin",
type = "percent", centraL = "Lc",
modelsupport = supp, criterion = "bic",
dropNA = TRUE,
reg.lim = c(17, 25, 35),
block.cols = cols)
# Color code regions by block (precaudal vs caudal):
blockcols <- list(PreCaudal = c("#BF812D","#8C510A","#543005"),
Caudal = c("#C7EAE5","#80CDC1","#35978F","#01665E","#236E30"))
plotvertmap(dolphin_pco, name = "Dolphin",
type = "percent", centraL = "Lc",
modelsupport = supp, criterion = "bic",
dropNA = TRUE,
reg.lim = c(17, 25, 35),
block.cols = blockcols, block.lim = 25)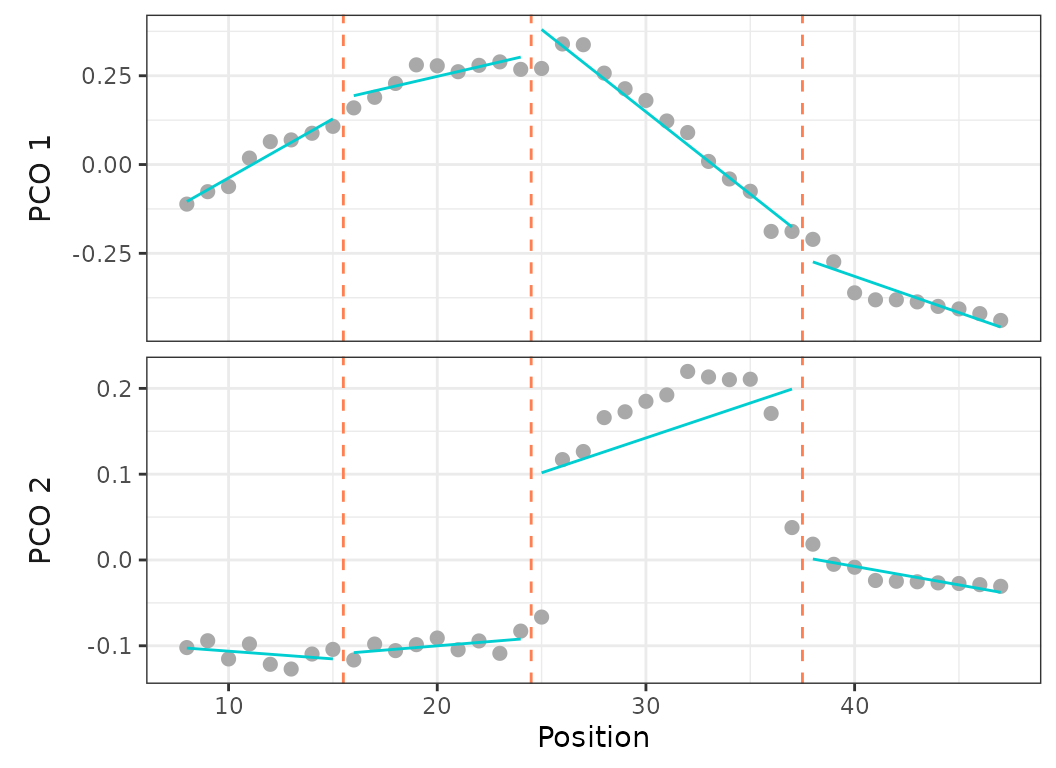
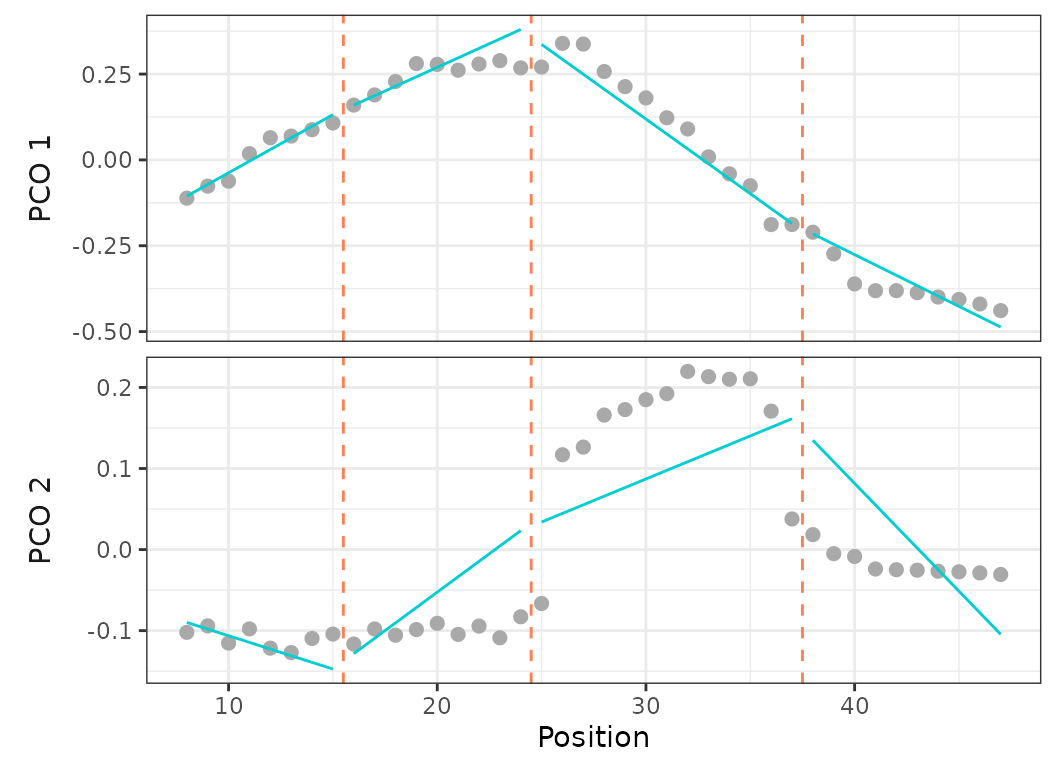
Fig.10. Vertebral maps with regions colored with a user-defined palette using viridis (top) and custom palettes for different blocks (bottom).
When coloring regions by blocks, regions spanning over more than one block will be allocated to the block in which the majority of its elements belong. In the example above, the 3rd region has one vertebra in the precaudal block and two in the caudal block, thus this region is considered as the 1st region in the caudal block.
Vertebral maps are created using ggplot2 meaning that they
can be modified similarly to ggplot plots. For instance, it
is possible to create a single plot with multiple vertebral maps with
the patchwork package.
# Run regions analysis on alligator and mouse data:
dat_alligator <- process_measurements(alligator, pos = 1)
pco_alligator <- svdPCO(dat_alligator, "gower")
regions_alligator <- calcregions(pco_alligator, scores = 1:3,
noregions = 5)
supp_alligator <- modelsupport(modelselect(regions_alligator))
dat_mouse <- process_measurements(musm, pos = 1)
pco_mouse <- svdPCO(dat_mouse, "gower")
regions_mouse <- calcregions(pco_mouse, scores = 1:3,
noregions = 5)
supp_mouse <- modelsupport(modelselect(regions_mouse))
# Create vertebral maps:
vertmaps <- list(alligator = plotvertmap(pco_alligator, name = "Alligator", centraL = "CL",
modelsupport = supp_alligator, type = "percent",
dropNA = TRUE),
mouse = plotvertmap(pco_mouse, name = "Mouse", centraL= "CL",
modelsupport = supp_mouse, type = "percent",
dropNA = TRUE),
dolphin = plotvertmap(dolphin_pco, name = "Dolphin", centraL= "Lc",
modelsupport = supp, type = "percent",
dropNA = TRUE))
# Remove x axis for plots except bottom one:
library(ggplot2)
for (i in seq_along(vertmaps)[-length(vertmaps)]){
vertmaps[[i]] <- vertmaps[[i]] + theme(axis.text.x = element_blank(),
axis.ticks.x = element_blank(),
axis.title.x = element_blank())
}
# Stack plots together:
library(patchwork)
wrap_plots(vertmaps) + plot_layout(ncol = 1, byrow = FALSE)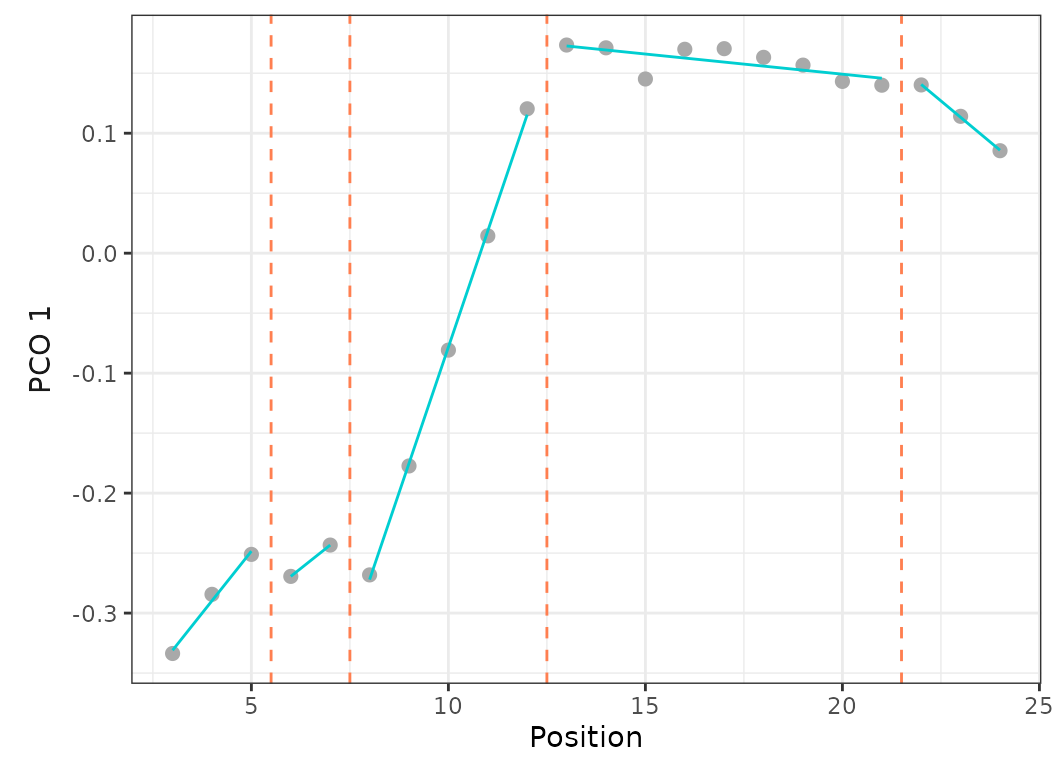
Fig.11. Vertebral maps of alligator, mouse, and dolphin stacked in a single figure.
Model fitting parameters
As briefly described in the section above, there are several different parameters available when fitting the segmented linear regressions.
Continuous vs. discontinuous fit
While the older regions package automatically fitted discontinuous segments, MorphoRegions allows users to choose between a continuous or discontinuous fit. In a continuous fit, the first point of the segment aligns with the last point of the preceding segment, modeling gradational variation without large jumps between segments. Because the continuous fit forces continuity between successive segments, it puts more constrains on the model fitting compared to a discontinuous fit. Depending on the dataset, one or the other fitting method might be appropriate. It’s advised to visually inspect the distributions of elements along the structure using the PCO scores that will be implemented in the analysis (as in Fig.2) before choosing an option.
# Plot a discontinuous model:
plotsegreg(dolphin_pco, scores = 1:2,
bps = c(15, 24, 37),
cont = FALSE)
# Plot a continuous model:
plotsegreg(dolphin_pco, scores = 1:2,
bps = c(15, 24, 37),
cont = TRUE)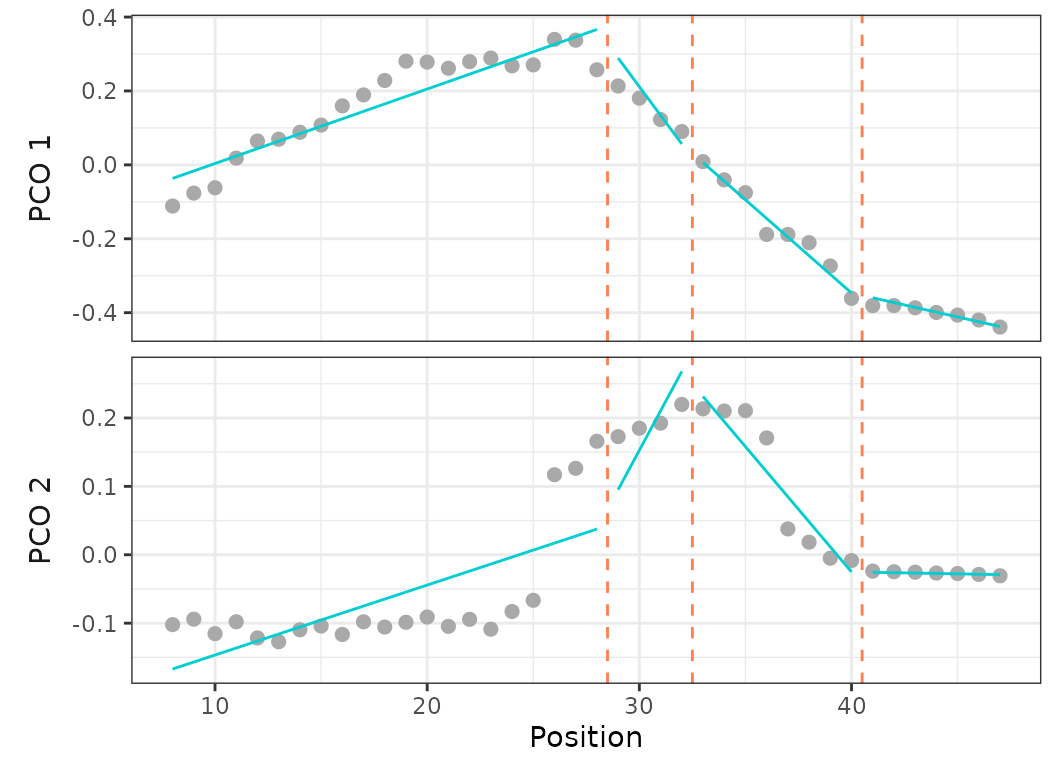
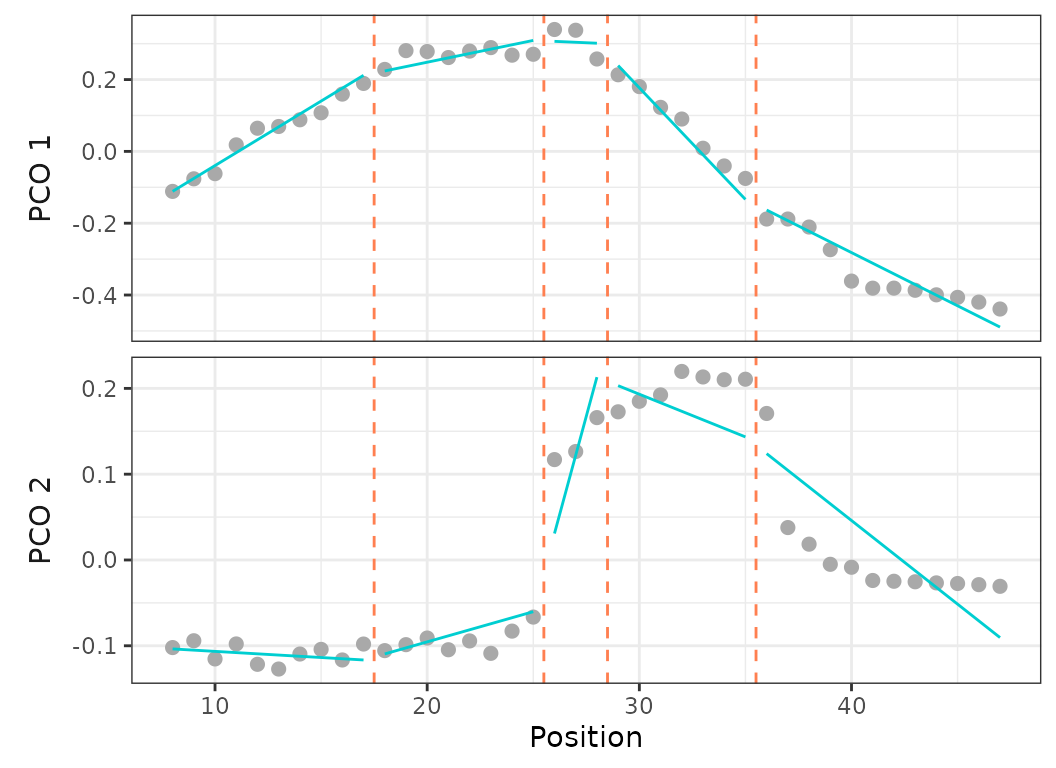
Fig.12. Best fit on PCOs 1 and 2 of a 4 region models with breakpoints at 15, 24, and 37, using a discontinuous model (left) and a continuous model (right).
Minimum number of elements per region
By default, the minimum number of elements (vertebrae) in each region
is set to 3. This parameter can, however, be changed with the
minvert argument in calcregions(). Note
that setting the minimum value to 2 might results in linear segments
made of only two points, which will have a perfect fit (RSS=0 for these
segments) and might artificially increase the goodness of fit.
# Fitting up to 5 regions for alligator data with 2 vertebrae minimum per segment:
reg.all.2 <- calcregions(pco_alligator, scores = 1,
minvert = 2, noregions = 5,
cont = FALSE)
plotsegreg(pco_alligator, scores = 1,
modelsupport = modelsupport(modelselect(reg.all.2)),
criterion = "bic")
# Fitting up to 5 regions for alligator data with 4 vertebrae minimum per segment:
reg.all.4 <- calcregions(pco_alligator, scores = 1,
minvert = 4, noregions = 5,
cont = FALSE)
plotsegreg(pco_alligator, scores = 1,
modelsupport = modelsupport(modelselect(reg.all.4)),
criterion = "bic")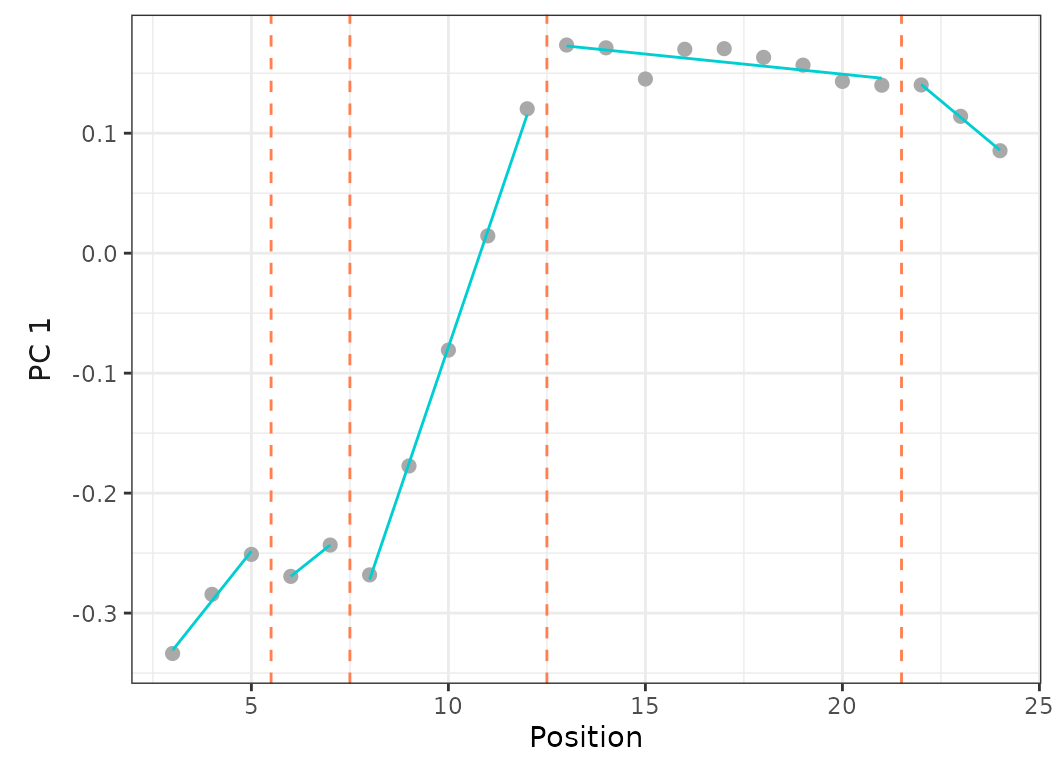
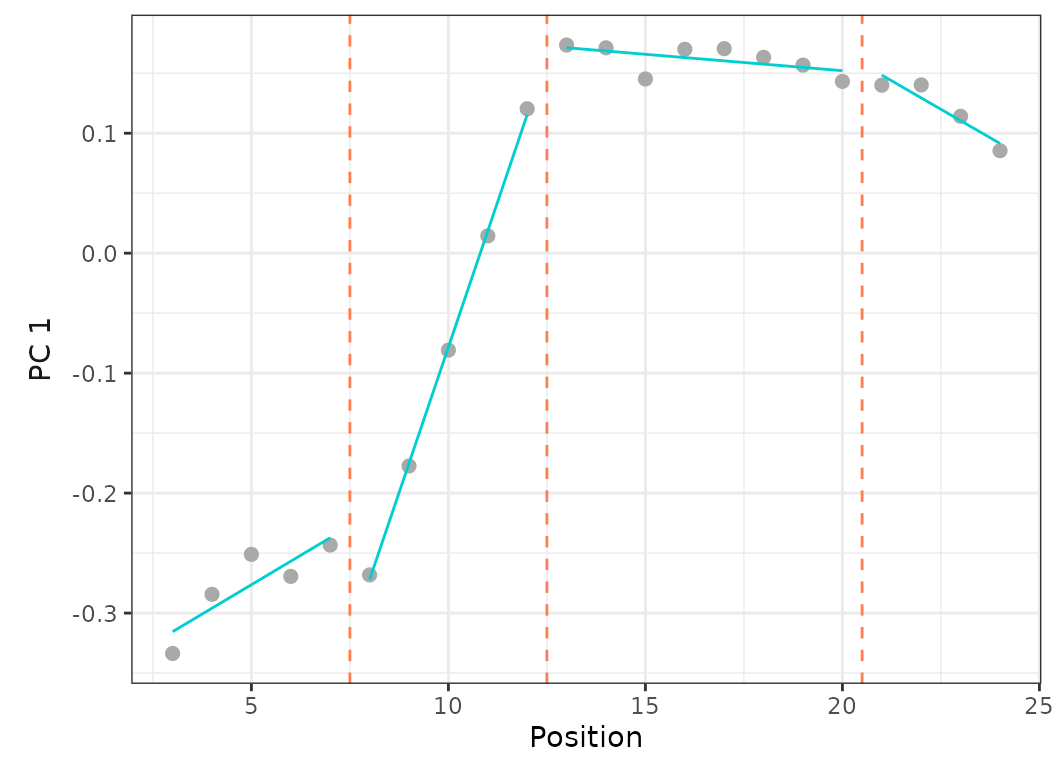
Fig.13. Best fit on PCO 1 of models up to 5 regions with a minimum of 2 vertebrae per region (left) or 4 vertebrae per region (right).
When setting the minimum number of vertebrae per region to 2, the best fit model (using BIC) is the 5 regions model with RSS = 0.001 and BIC weight = 0.99.
When forcing the minimum number of vertebrae per region to 4, the best fit model (using BIC) is the 4 regions model with RSS = 0.002 and BIC weight = 0.61. The 5 regions model is only the 3rd best model (after the 3 regions model) with RSS = 0.002 and BIC weight = 0.02.
Forcing or preventing specific breakpoint positions
The omitbp and includebp arguments of
calcregions allows one to prevent or force breakpoint(s) at
specific position(s), respectively.
# Fit models for dolphin preventing any BP in the thoracic and lumbar regions:
reg.dol.noBP <- calcregions(dolphin_pco, scores = PCOs, noregions = 5,
omitbp = 8:25)
plotsegreg(dolphin_pco, scores = PCOs,
modelsupport = modelsupport(modelselect(reg.dol.noBP)),
criterion = "bic")
# Fit models for dolphin forcing BPs at anatomical region boundaries:
reg.dol.forceBP <- calcregions(dolphin_pco, scores = PCOs, noregions = 5,
includebp = c(17, 25, 35))
plotsegreg(dolphin_pco, scores = PCOs,
modelsupport = modelsupport(modelselect(reg.dol.forceBP)),
criterion = "bic")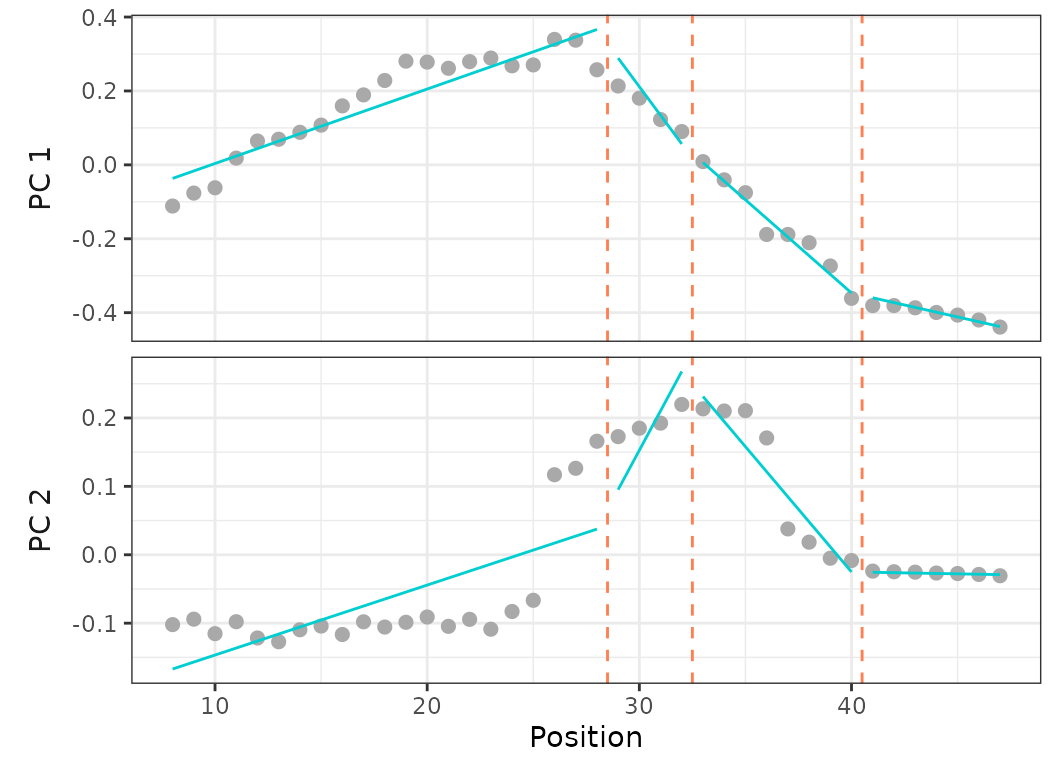
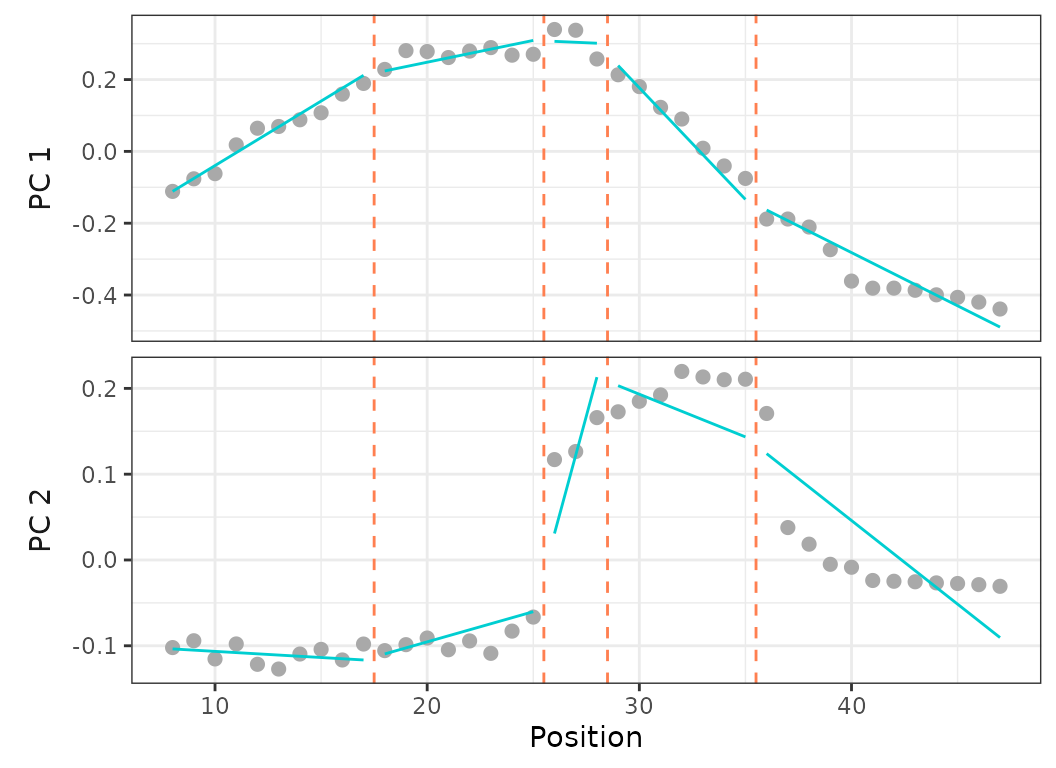
Fig.14. Best fit on PCO 1 and 2 of models up to 5 regions preventing any breakpoint in the thoraco-lumbar region (vertebrae 8 to 25) (top) and forcing breakpoints at anatomical region boundaries (vertebrae 17, 25, 35) (bottom).
Exhaustive vs. non-exhaustive search
MorphoRegions has been implemented to fit numerous models
efficiently enough to run in a few minutes on most computers. However,
in the case of quite complex models (numerous elements, regions to fit,
and/or PCO scores), computational time can quickly increase. To speed up
the process, it is possible to fit models in a non-exhaustive way by
setting exhaus = FALSE in calcregions().
When a non-exhaustive search is used, all possible models for 1 and 2 regions models are fitted, but for 3 and more regions, the algorithm will only fit a subset of all the possible models. For a given number of regions, , the optimal positions of breakpoints of the models with regions are used as a starting point to define the subset of models to fit.
reg.dol.Nex <- calcregions(dolphin_pco, scores = PCOs, noregions = 5,
exhaus = FALSE)
summary(reg.dol.Nex)
#> Regions Possible Tested Saved Comp. method Saving method
#> 1 1 1 1 Exhaustive All
#> 2 35 35 35 Exhaustive All
#> 3 528 528 115 Non-exhaus SD/2
#> 4 4495 3220 421 Non-exhaus SD/2
#> 5 23751 11648 424 Non-exhaus SD/2In a non-exhaustive search, using summary() the
calcregions() output returns, for each number of regions,
the total number of possible models, the number of models actually
fitted, and the number of models saved to output in order to limit
memory usage.
If the variability of breakpoint positions is calculated on non-exhaustive search output, the proportion of best models used to calculate the variability corresponds to the given percentage of all possible models. It’s therefore possible that in some cases the number of models saved by the non-exhaustive search is lower than the requested percentage. In such cases, the variability will be calculated on all saved models and a warning message is returned.
calcBPvar(reg.dol.Nex, noregions = 5, pct = 0.1,
criterion = "bic")
#> Warning in calcBPvar(reg.dol.Nex, noregions = 5, pct = 0.1, criterion = "bic"): Number of models provided lower than percentage requested. Weighted means and
#> SD calculated on 1.79% of total number of models.
#> BP 1 BP 2 BP 3 BP 4
#> wMean 23.154 27.354 33.985 39.722
#> wSD 0.524 0.509 0.677 0.630
#> - Computed using top 1.79% of modelsBecause average and variability of breakpoint position is calculated using weighted means and SD, respectively, results usually remain similar to those obtained using full top 10% (models fitted with an exhaustive search) unless there are numerous models which fit the data well.
calcBPvar(regionresults, noregions = 5, pct = 0.1,
criterion = "bic")
#> BP 1 BP 2 BP 3 BP 4
#> wMean 23.154 27.354 33.985 39.722
#> wSD 0.524 0.509 0.678 0.630
#> - Computed using top 10% of modelsParallel computing
For specimens with a high number of elements and/or numerous regions
and for which computing non-exhaustive searches are still time
consuming, the cl argument in calcregions()
allows one to implement parallel computing. See the cl
argument to pbapply::pblapply() for more details on how it
can be specified. Below, we request 6 cores to be used (supplying a
number is not available on Windows).
# Exhaustive search with parallel computing using 6 cores:
calcregions(dolphin_pco, scores = 2, noregions = 10, exhaus = TRUE,
cl = 6)Note that parallel computing will only be significantly faster on heavy computing specimens.
Intraspecific variability
To account for variability within a species or group, it is possible to run the regionalization analysis on multiple specimens in a single analysis, both for traditional morphometric and geometric morphometric datasets.
Raw morphometrics (Pipeline 1):
The porpoise dataset contains linear measurements on the
vertebrae of 3 different specimens of harbor porpoise.
data("porpoise")| Elements | Variables | |
|---|---|---|
| Porpoise 1 | 58 | 17 |
| Porpoise 2 | 56 | 17 |
| Porpoise 3 | 59 | 17 |
The total number of elements (vertebrae) can differ among specimens, but the number of variables and their order must be the same. The highest number of elements is used to generate the vertebral map. In the porpoise dataset, the first specimen has 58 post-cervical vertebrae, the second has 56, and the third one has 59, but they all have 17 variables. In the case of differing number of elements, homologous elements (vertebrae) must be identified by the same number in the positional information column of the data.
# Process measurements data
porpoise_data <- process_measurements(list(porpoise1,
porpoise2,
porpoise3),
pos = "Vertebra")
# Compute PCOs
porpoise_pco <- svdPCO(porpoise_data, "gower")
# Plot PCOs
plot(porpoise_pco, 1:2)
plot(porpoise_pco, pc_x = 1, pc_y = 2)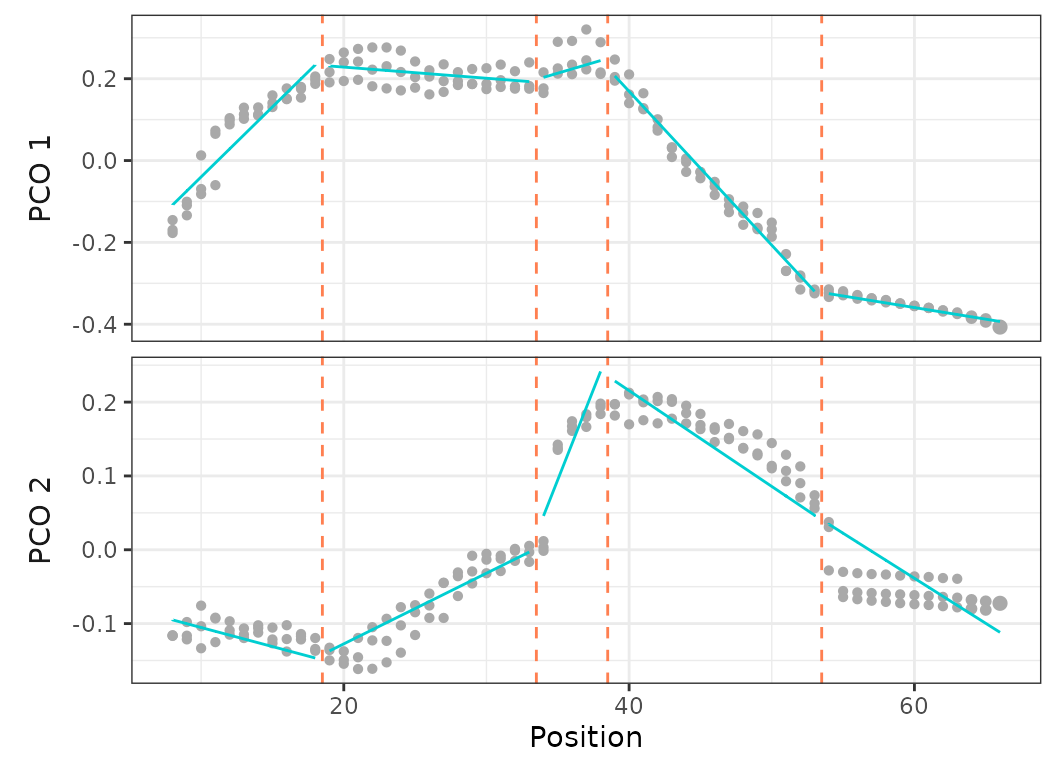
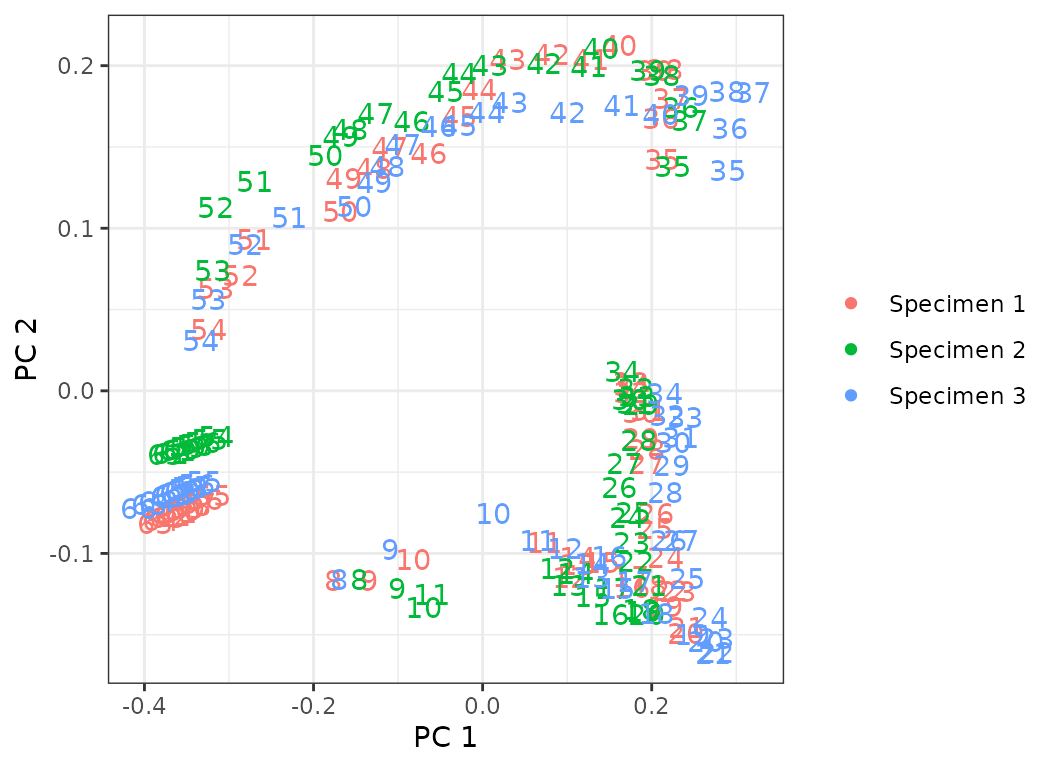
Fig.15. PCO plots of the three specimens of harbor porpoise.
PCs from traditional morphometrics (Pipeline 2):
Using the two first specimens from the porpoise dataset,
we first need to compute a PCA and then use the PC scores as input data
for process_PC().
# Combine data of porpoises 1 and 2:
porpoise_dat <- rbind(porpoise1, porpoise2)
# Compute PCA on both specimens together:
PCA_porp <- prcomp(porpoise_dat, scale = T, center = T)
# Extract PC scores and eigenvalues:
PCscores_porp <- PCA_porp$x
PCeigen_porp <- PCA_porp$sdev^2In addition to the PC scores and eigenvalues, the raw data needs to
be provided as a list where each element of the list contains the raw
data of a specimen. The positional information of the raw data can be
provided using the pos argument. Similarly, the positional
information in the PC score dataset can be provided with the
pocPC argument. The positional information for
pocPC needs to be provided as a named list
where each element of the list contains positional information for a
specimen, and names correspond to specimen names. In such case, the list
containing raw data must also be a named list using the
same specimen names as in the posPC list. Note that
providing positional information is particularly important if different
vertebrae have been sampled in different specimens.
# Create named list of raw data:
porpoises <- list(porpoise1, porpoise2)
names(porpoises) <- c('Porpoise1', 'Porpoise2')
# Create list of positional information:
posPCporp <- lapply(porpoises,'[[',1)
posPCporp
#> $Porpoise1
#> [1] 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32
#> [26] 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57
#> [51] 58 59 60 61 62 63 64 65
#>
#> $Porpoise2
#> [1] 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32
#> [26] 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57
#> [51] 58 59 60 61 62 63
# Input data in MorphoRegions:
porpoise_pca <- process_PC(data = porpoises, pos = 'Vertebra',
pcscores = PCscores_porp, eigenvals = PCeigen_porp,
posPC = posPCporp)
plot(porpoise_pca, pc_x = 1, pc_y = 2)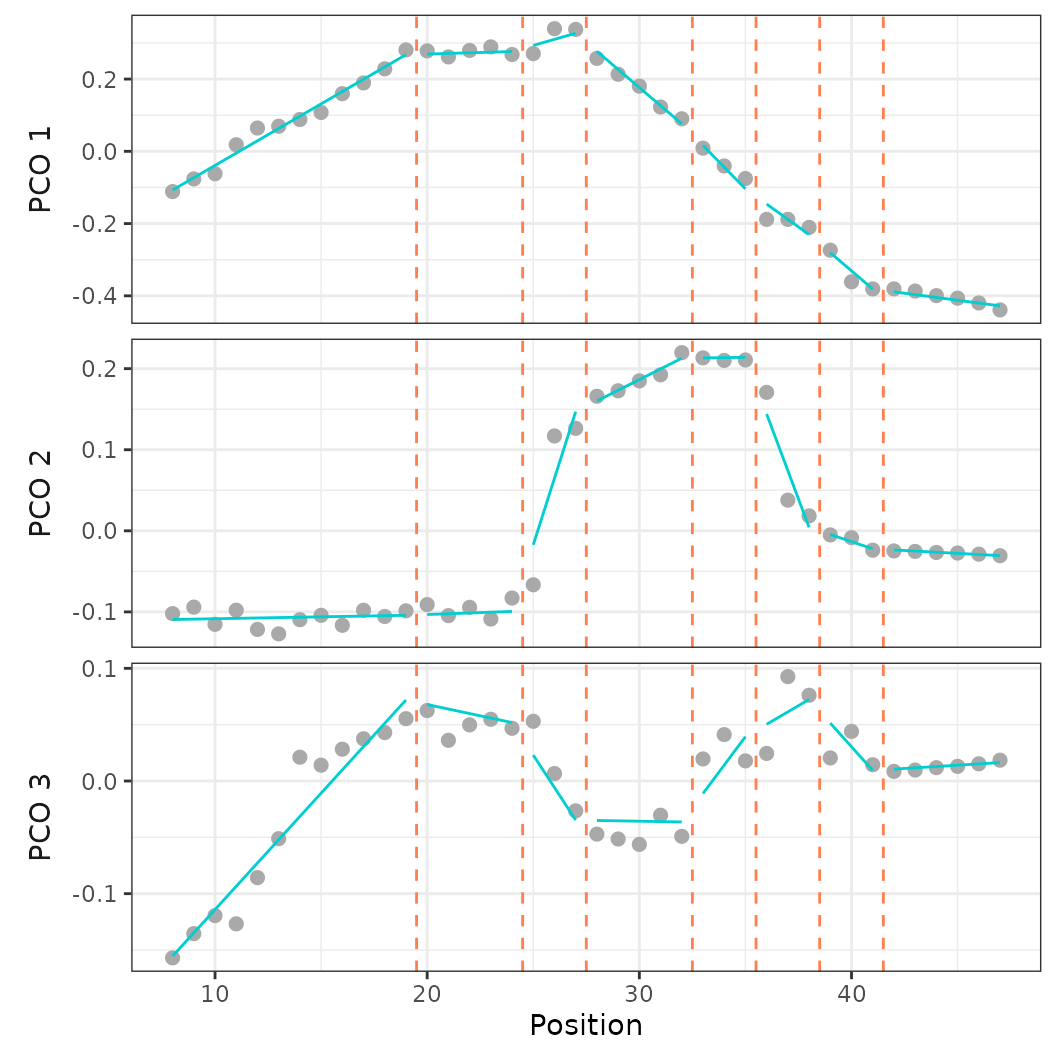
Fig.16. Morphospace of two specimens of harbor porpoise.
PCs from geometric morphometrics (Pipeline 3):
The wolves_gmm dataset contains 3D geometric
morphometric data and PC scores of 10 subsampled vertebrae in two
specimens of wolves. For multiple specimens, specimen names must be
provided as a vector of equal length to the total number of vertebrae in
the sample (and in the same order as in the landmark array and the PC
score dataset).
data("wolves_gmm")
names(wolves_gmm)
#> [1] "coord_gpa" "specimens" "vertebrae" "scores" "eigenvals"
# Positional information:
wolves_gmm$vertebrae
#> [1] 3 5 7 8 14 17 20 21 24 27 3 5 7 8 14 17 20 21 24 27
# Specimen information:
wolves_gmm$specimens
#> [1] "MU071" "MU071" "MU071" "MU071" "MU071" "MU071" "MU071" "MU071" "MU071"
#> [10] "MU071" "MU073" "MU073" "MU073" "MU073" "MU073" "MU073" "MU073" "MU073"
#> [19] "MU073" "MU073"The data can then be converted to regions_pco object
with the process_gmPC() function (as for a single
specimen).
wolf_pca <- process_gmPC(data = wolves_gmm$coord_gpa,
pcscores = wolves_gmm$scores,
eigenvals = wolves_gmm$eigenvals,
pos = wolves_gmm$vertebrae,
specimens = wolves_gmm$specimens)Perform regionalization analysis:
Once data of each specimen have been converted to
regions_pco object (either through pipeline 1, 2, or 3),
the process to run the analysis is similar to running the
analysis on a single specimen.
# Calculate number of PCOs to use in subsequent analyses
PCOs <- PCOselect(porpoise_pco, "variance", cutoff = .05)
# Performance for all models across all breakpoint combos
regionresults <- calcregions(porpoise_pco, scores = PCOs,
noregions = 3)
# Adding more models to existing results
regionresults <- addregions(regionresults, noregions = 4:5,
exhaus = FALSE)
# Select best model
models <- modelselect(regionresults)
supp <- modelsupport(models)
supp
#> - Model support (AICc)
#> Regions BP 1 BP 2 BP 3 BP 4 sumRSS AICc deltaAIC model_lik Ak_weight
#> 5 18 33 38 53 0.105 -2769.304 0.000 1 1
#> 4 20 40 52 . 0.148 -2656.161 113.142 0 0
#> 3 21 39 . . 0.293 -2426.584 342.720 0 0
#> 2 37 . . . 0.631 -2167.924 601.380 0 0
#> 1 . . . . 2.155 -1749.033 1020.271 0 0
#> Region score: 5
#>
#> - Model support (BIC)
#> Regions BP 1 BP 2 BP 3 BP 4 sumRSS BIC deltaBIC model_lik BIC_weight
#> 5 18 33 38 53 0.105 -2709.414 0.000 1 1
#> 4 20 40 52 . 0.148 -2607.254 102.160 0 0
#> 3 21 39 . . 0.293 -2388.776 320.638 0 0
#> 2 37 . . . 0.631 -2141.330 568.084 0 0
#> 1 . . . . 2.155 -1733.764 975.650 0 0
#> Region score: 5
# Calculate variability of optimal breakpoints
bpvar <- calcBPvar(regionresults, noregions = 5, pct = .05,
criterion = "bic")
bpvar
#> BP 1 BP 2 BP 3 BP 4
#> wMean 18.014 33.124 37.695 52.655
#> wSD 0.651 0.510 0.461 0.493
#> - Computed using top 4.57% of models
# Calculate model performance (R2) of best model
modelperf(porpoise_pco, scores = PCOs, modelsupport = supp,
criterion = "bic", model = 1)
#> Breakpoints: 18, 33, 38, 53
#>
#> - Univariate:
#> R² Adj. R²
#> PCO.1 0.984 0.983
#> PCO.2 0.936 0.935
#>
#> - Multivariate:
#> R² Adj. R²
#> 0.974 0.974
# Plot PCOs of best fit model
plotsegreg(porpoise_pco, scores = 1:2, modelsupport = supp,
criterion = "bic", model = 1)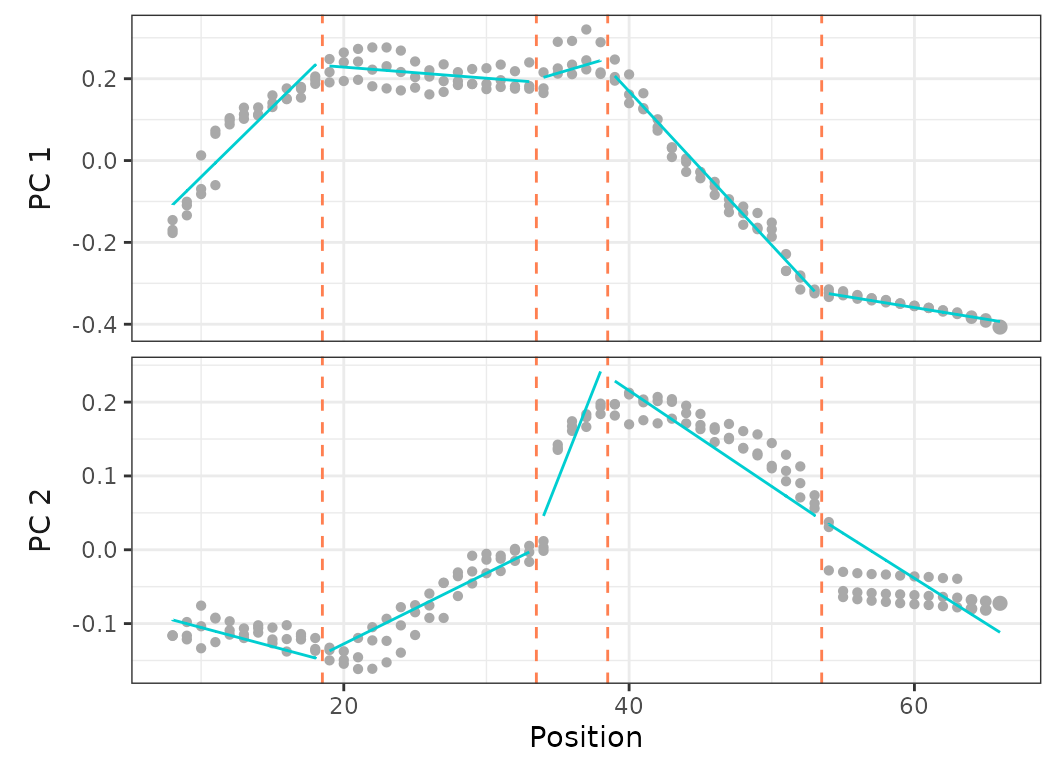
Fig.18. Best fit model (based on BIC) on PCO 1 and 2 calculated on the three harbor porpoise specimens.
# Plot vertebral map of best fit model
plotvertmap(porpoise_pco, name = "Porpoise",
type = "percent", centraL = "Lc",
reg.lim = c(20, 34, 54), dropNA = TRUE,
bpvar = bpvar, sd.col = "grey20")
Fig.19. Vertebral map of best fit model (including variance in breakpoint position) based on the three harbor porpoise specimens.
Other useful options
Fitting a specific model
Instead of fitting all possible models for a given range of region
number, calcmodel() allows one to fit a single model with
user-defined breakpoint positions.
#Fit a single breakpoints model
regionsmodel <- calcmodel(dolphin_pco, scores = 1:3, cont = TRUE,
bps = c(19, 24, 27, 32, 35, 38, 41))
regionsmodel
#> Regions BP 1 BP 2 BP 3 BP 4 BP 5 BP 6 BP 7 sumRSS RSS.1 RSS.2 RSS.3
#> 8 19 24 27 32 35 38 41 0.034 0.01 0.01 0.013
#>
#> - Support:
#> AICc BIC
#> -885.1162 -818.3415
#Evaluate performance (R2) on that model
modelperf(regionsmodel)
#> Breakpoints: 19, 24, 27, 32, 35, 38, 41
#>
#> - Univariate:
#> R² Adj. R²
#> PCO.1 0.996 0.995
#> PCO.2 0.983 0.978
#> PCO.3 0.904 0.879
#>
#> - Multivariate:
#> R² Adj. R²
#> 0.990 0.987
#Plot model results:
plotsegreg(regionsmodel, scores = 1:3)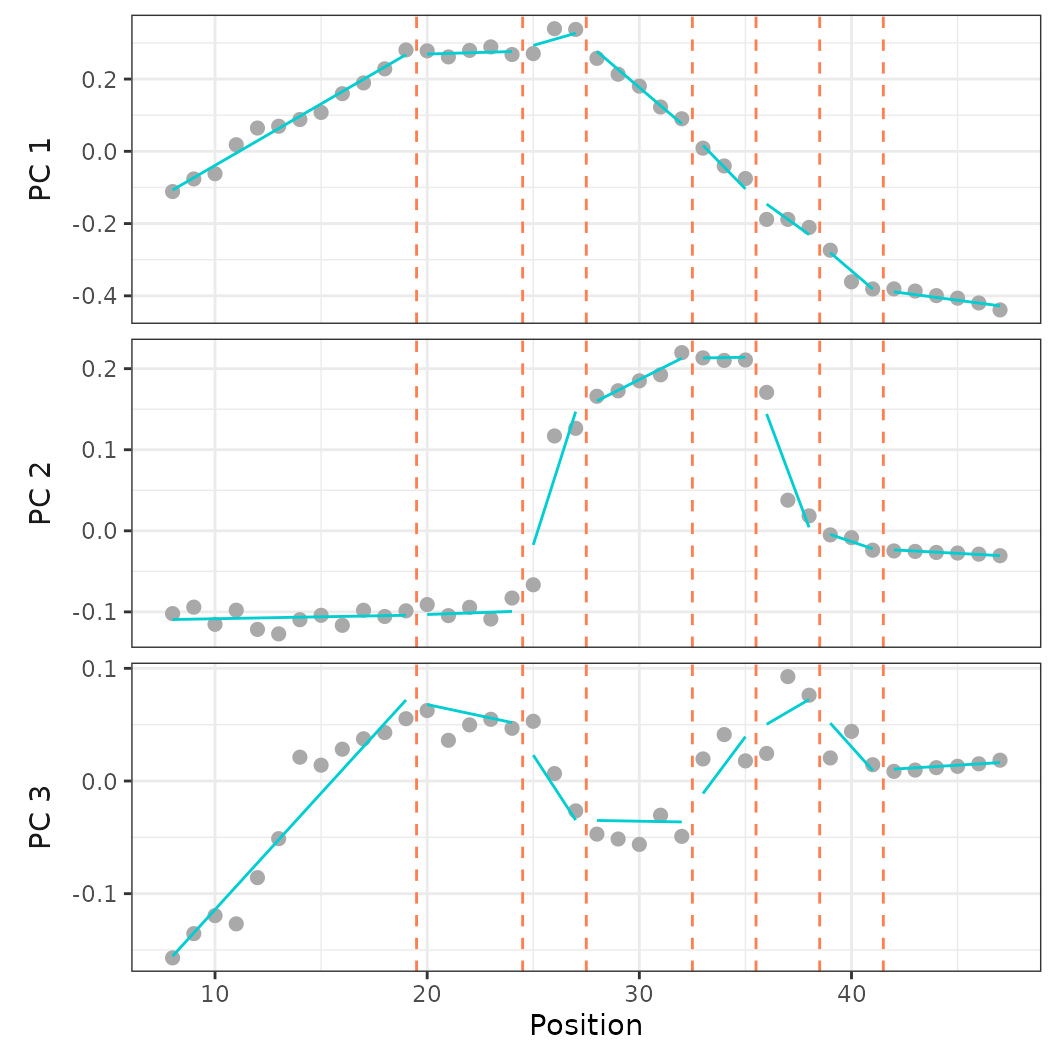
Fig.20. Best fit segmented linear regression model on PCO 1-3 when forcing breakpoint position at vertebrae 19, 24, 27, 32, 35, 38, and 41.
Subsampling elements
For specimens with numerous elements (more than 100), computational time can become high. It can then be useful to run the regionalization analysis on a subset of elements If the analyses are run in a comparative framework and there is substantial variation of the number of elements across specimens, it is also advised to subsample elements to a constant number of elements for all specimens.
subsample() allows one to subsample elements either by
providing the number of elements to retain or the proportion of total
elements to retain.
# Retain 33 vertebrae
dolphin_pco_sub.33 <- subsample(dolphin_pco, sample = 33)
# Retain 70% of initial number of vertebrae
dolphin_pco_sub.7 <- subsample(dolphin_pco, sample = .7)
plotvertmap(dolphin_pco_sub.33, name = "Dolphin 33")
plotvertmap(dolphin_pco_sub.7, name = "Dolphin .7")

Fig.21. Vertebral map of dolphin dataset with 33 subsampled vetrebrae (top) and 70% of total vertebrae subsampled (bottom). Vertebrae dropped are in grey and vertebrae retained are in blue shade*.
By default, elements to drop will be selected to be equidistant along
the structure (rounded to integers). A random subsampling of elements
along the structure can be applied by supplying
type = "random" to subsample().
Simulating data
simregions() allows one to simulate datasets that
satisfy some user-defined constraints such as number of elements,
regions, PCO axes, minimum number of elements per region, variance
around the linear fits, and difference in slope between successive
linear segments.
# Simulate data
set.seed(2024)
sim <- simregions(nvert = 40, nregions = 5, nvar = 4, r2 = .92,
minvert = 3, cont = TRUE)
# Plot:
plot(sim, scores = 1:2)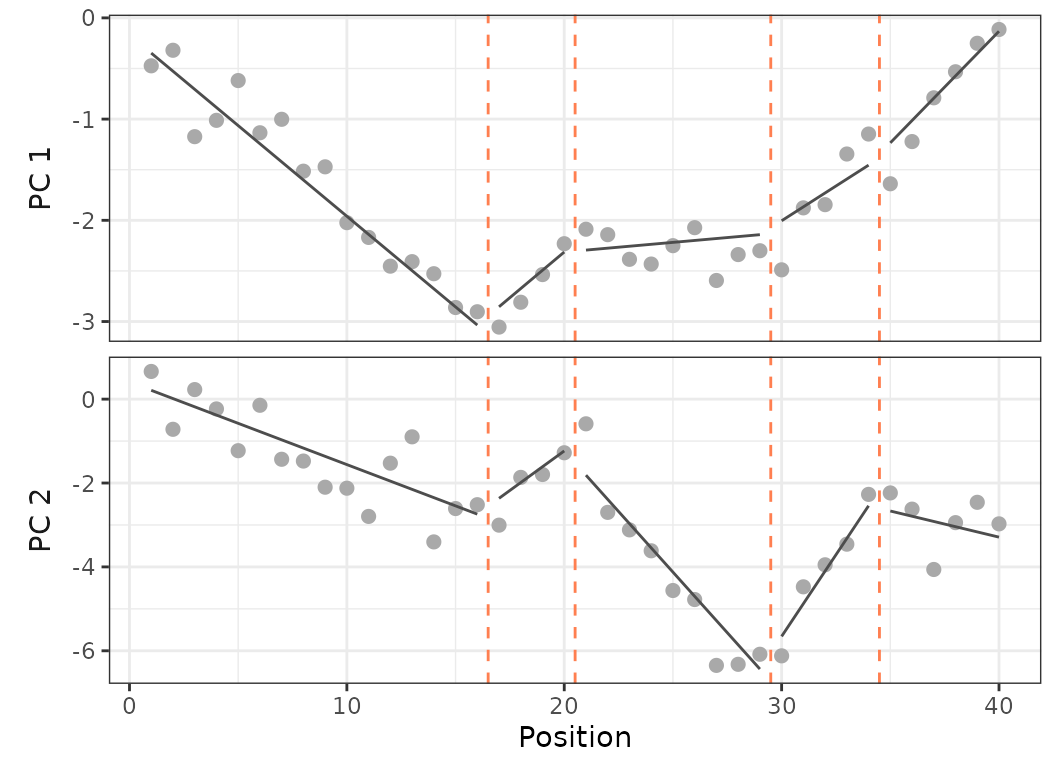
Fig.22. Plot on PCO 1 and 2 of the simulated dataset with 40 vertebrae and 5 regions (4 breakpoints).
The regionalization analysis can then be run on the simulated dataset.
simresults <- calcregions(sim, scores = 1:4, noregions = 6)
simmodels <- modelselect(simresults)
simmodels
#> Regions BP 1 BP 2 BP 3 BP 4 BP 5 sumRSS RSS.1 RSS.2 RSS.3 RSS.4
#> 1 . . . . . 239.092 26.450 72.184 28.139 112.318
#> 2 29 . . . . 185.535 8.393 39.551 26.728 110.863
#> 3 19 28 . . . 148.383 4.114 31.513 22.545 90.211
#> 4 20 28 34 . . 128.804 4.623 23.291 9.623 91.267
#> 5 15 21 28 34 . 109.794 1.835 11.249 9.891 86.820
#> 6 4 15 21 28 34 105.817 1.724 11.209 7.410 85.474
simsupport <- modelsupport(simmodels)
simsupport
#> - Model support (AICc)
#> Regions BP 1 BP 2 BP 3 BP 4 BP 5 sumRSS AICc deltaAIC model_lik Ak_weight
#> 5 15 21 28 34 . 109.794 8.147 0.000 1.000 0.988
#> 6 4 15 21 28 34 105.817 17.655 9.508 0.009 0.009
#> 4 20 28 34 . . 128.804 19.417 11.270 0.004 0.004
#> 3 19 28 . . . 148.383 28.790 20.644 0.000 0.000
#> 2 29 . . . . 185.535 52.184 44.037 0.000 0.000
#> 1 . . . . . 239.092 81.221 73.075 0.000 0.000
#> Region score: 5
#>
#> - Model support (BIC)
#> Regions BP 1 BP 2 BP 3 BP 4 BP 5 sumRSS BIC deltaBIC model_lik BIC_weight
#> 3 19 28 . . . 148.383 79.292 0.000 1.000 0.650
#> 5 15 21 28 34 . 109.794 81.855 2.562 0.278 0.181
#> 4 20 28 34 . . 128.804 82.028 2.736 0.255 0.166
#> 2 29 . . . . 185.535 89.668 10.376 0.006 0.004
#> 6 4 15 21 28 34 105.817 101.326 22.034 0.000 0.000
#> 1 . . . . . 239.092 104.869 25.577 0.000 0.000
#> Region score: 3.52
modelperf(sim, modelsupport = simsupport,
criterion = "aic", model = 1)
#> Breakpoints: 15, 21, 28, 34
#>
#> - Univariate:
#> R² Adj. R²
#> PCO.1 0.931 0.921
#> PCO.2 0.907 0.894
#> PCO.3 0.883 0.866
#> PCO.4 0.884 0.867
#>
#> - Multivariate:
#> R² Adj. R²
#> 0.888 0.872
#Plot best model fit
plotsegreg(sim, scores = 1:2, modelsupport = simsupport,
criterion = "aic", model = 1)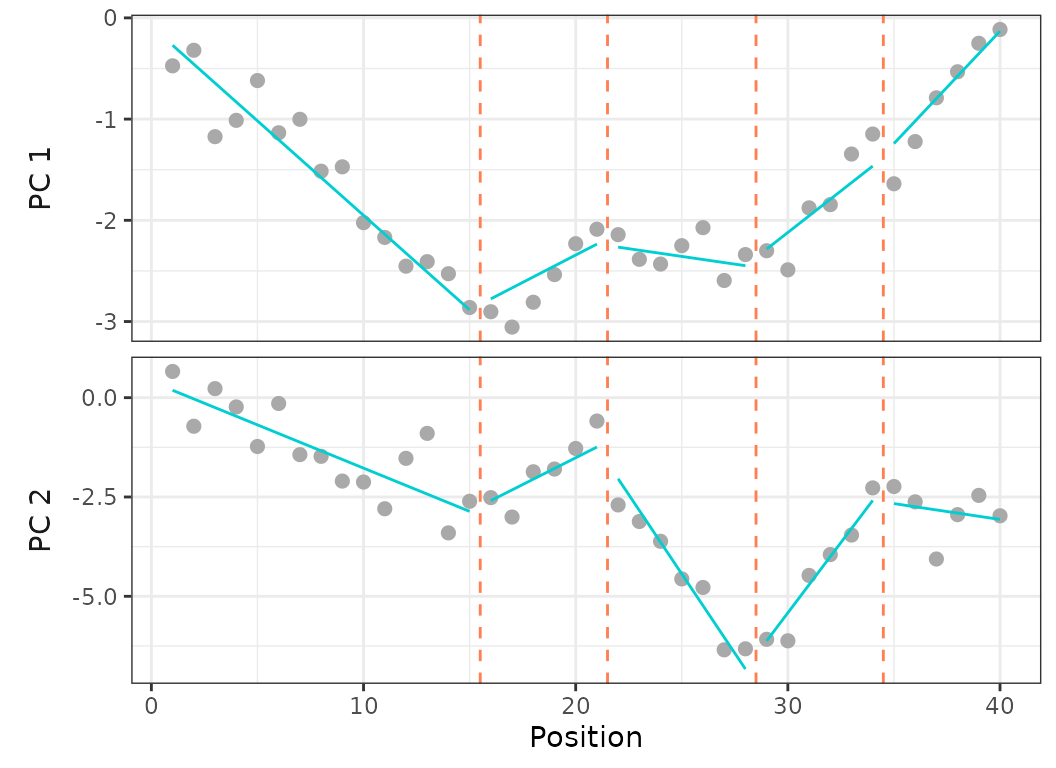
Fig.23. Plot on PCO 1 and 2 of the recovered best fit on regionalization analysis ran on the simulated dataset.
The simulated dataset had 5 predefined regions with breakpoints at vertebrae 16, 20, 29, and 34. When running the regionalization analysis on the simulated dataset, the analysis correctly recovered the 5 regions model as the best fit model using AICc, however the 5 regions model is second best using BIC.
The AICc best fit model recovered breakpoints at vertebrae 15, 21, 28, 34. The recovered position of the first three breakpoints differ sightly from the simulated positions, and the recovered position of the fourth breakpoint matches that of the simulated data.
# Simulated data breakpoints:
sim$BPs
#> breakpoint1 breakpoint2 breakpoint3 breakpoint4
#> 16 20 29 34
# Recovered best fit breakpoints:
simsupport$Model_support[1, 2:5]
#> breakpoint1 breakpoint2 breakpoint3 breakpoint4
#> 26746 15 21 28 34
# Variability of recovered best fit breakpoints:
calcBPvar(simresults, noregions = 5, pct = 0.1,
criterion = "aic")
#> BP 1 BP 2 BP 3 BP 4
#> wMean 15.184 20.652 28.138 34.249
#> wSD 1.618 0.788 0.511 0.710
#> - Computed using top 10% of models